Subido por
Felipe de Jesús Cortes Delgadillo
Registro Sanitario Dispositivos Médicos: Guía COFEPRIS
Anuncio
REQUISITOS PARA EL TRÁMITE DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS Homoclaves: COFEPRIS-04-001-A, COFEPRIS-04-001-B y COFEPRIS-04-001-C Dispositivos Médicos Clase I conforme al Art. 83 del Reglamento de Insumos para la Salud COFEPRIS C COFEPRIS C SALUD PROTECCION COMISION LA PROTECCIÓN FEDERAL PARA LA COMISIÓN FEDERAL SALUD SECRETARÍA SECRETARIA DE SALUD fM SANITARIOS RIESCOS SANITARIOS CONTRA CONTRA DIESCOS Ο D gob.mx/cofepris ob.mx/cofepris Tabla de contenido 1. Documentos que se recomienda consultar-------------------------------------------------------------2 2. Acrónimos------------------------------------------------------------------------------------------------------2 3. Objetivo---------------------------------------------------------------------------------------------------------3 4. Alcance---------------------------------------------------------------------------------------------------------3 5. Introducción---------------------------------------------------------------------------------------------------3 6. Concepto de Dispositivo Médico-------------------------------------------------------------------------4 7. Etapas del proceso de obtención del Registro Sanitario para dispositivos médicos Clase I, II, y III.-------------------------------------------------------------------------5 7.1 ¿El dispositivo médico requiere de registro sanitario?---------------------------------------------6 7.2. Identificación de la categoría de uso del dispositivo médico.-----------------------------------6 7.3. Clasificación de los dispositivos médicos de acuerdo con el riesgo sanitario----------------9 7.4. Identificación de Homoclave y Modalidad-----------------------------------------------------------10 7.5. Requisitos legales, administrativos y técnicos para el trámite de Registro Sanitario-------11 8. Referencias bibliográficas----------------------------------------------------------------------------------12 9. Checklist Dispositivos Médicos Clase I-------------------------------------------------------------------13 10. Checklist Dispositivos Médicos Clase II-----------------------------------------------------------------24 11. Checklist Dispositivos Médicos Clase III-----------------------------------------------------------------37 12. Checklist Software como Dispositivo Médico (ScDM) Clase I, ll y III------------------------------52 13. Consideraciones generales--------------------------------------------------------------------------------60 14. Notas ScDM---------------------------------------------------------------------------------------------------61 1. Documentos que se recomienda consultar Guía para el ingreso de trámites. COFEPRIS. 2020. [Recurso electrónico]. https://www.gob.mx/cms/uploads/attachment/file/535715/Guia_Ingreso_Tramites_Febrero_2020.pdf Instructivo de llenado del formato Autorizaciones, Certificados y Visitas. [Recurso electrónico]. COFEPRIS https://www.gob.mx/cms/uploads/attachment/file/348649/Instructivo_Autorizaciones.pdf 2. Acrónimos COFEPRIS: Comisión Federal para la Protección contra Riesgos Sanitarios. DM: Dispositivos Médicos. RIS: Reglamento de Insumos para la Salud. LGS: Ley General de Salud. LFPA: Ley Federal de Procedimiento Administrativo. LFD: Ley Federal de Derechos. FEUM: Farmacopea de los Estados Unidos Mexicanos Suplemento para Dispositivos Médicos. 3. Objetivo Establecer los requisitos indispensables que deben cumplir los dispositivos médicos Clase I, II y III para la obtención del Registro Sanitario, y facilitar la integración de información en el dossier. 4. Alcance Esta guía está dirigida a toda persona física o moral que promuevan trámites de solicitud de registro sanitario de Dispositivos Médicos. 5. Introducción La regulación sanitaria se define como el conjunto de acciones preventivas que lleva a cabo el gobierno para normar y controlar las condiciones sanitarias, los establecimientos, las actividades, los productos, los equipos, los 15 vehículos y las personas que puedan representar riesgo o daño a la salud de la población en general. 15 En términos generales la regulación sanitaria de los dispositivos médicos (DM) debe encontrar un balance entre los siguientes objetivos: 1. Asegurar que los dispositivos médicos son clínicamente seguros, efectivos y eficaces. 2. Facilitar el acceso oportuno y equitativo de los pacientes a la tecnología en salud existente. 3. Fomentar el desarrollo continuo de innovaciones tecnológicas. Para la comercialización, fabricación y uso de los Dispositivos Médicos se requiere cumplir con ciertas autorizaciones sanitarias que garanticen que estos dispositivos son seguros y eficaces, según sea el caso, expedidas porpor la Comisión Federal parapara la la la Comisión Federal 15 Protección contra Riesgos Sanitarios (COFEPRIS). Protección contra Riesgos Sanitarios (COFEPRIS). La COFEPRIS es un órgano desconcentrado de la Secretaría de Salud con autonomía administrativa, técnica y operativa, la cual otorga el Registro Sanitario; autorización sanitaria con la cual un dispositivo médico nacional o extranjero puede ser distribuido y puesto en venta en territorio nacional. 3 En los artículos 179, 180, 181 y 182 del Reglamento de Insumos para la Salud (RIS) se establecen los requisitos que deben cumplir los DM para la obtención del Registro Sanitario, en ellos se incluye información científica, técnica y legal con la cual va a demostrar su seguridad y eficacia. A partir del 2005, estos registros cuentan con vigencia de 5 años, por lo que deberán ser prorrogados cumpliendo con las disposiciones que la Secretaría de Salud establezca. 15 En materia de regulación sanitaria específicamente de dispositivos médicos, en México se cuenta con el siguiente marco legal: Ley General de Salud (LGS). Ley Federal de Procedimiento Administrativo (LFPA). Reglamento de la Ley General de Salud en Materia de investigación para la salud. Reglamento de Insumos para la Salud (RIS). Normas oficiales mexicanas de DM. Cuando no se cuentan con normas nacionales se toman como referencia normas internacionales expedidas por la Organización Internacional para la Estandarización (ISO, por sus siglas en inglés), la Comisión Internacional Electrotécnica (IEC, por sus siglas en inglés), la Comisión Federal de Comunicaciones (FCC, por sus siglas en inglés) entre otras. Farmacopea de los Estados Unidos Mexicanos. Suplemento para Dispositivos Médicos 5.0. 6. Concepto de Dispositivo Médico Al instrumento, aparato, utensilio, máquina, software, producto o material implantable, agente de diagnóstico, material, sustancia o producto similar, para ser empleado, solo o en combinación, directa o indirectamente en seres humanos; con alguna(s) de las siguientes finalidades de uso: 13,14 Diagnóstico, prevención, vigilancia o monitoreo, y/o auxiliar en el tratamiento de enfermedades; Diagnóstico, vigilancia o monitoreo, tratamiento, protección, absorción, drenaje, o auxiliar en la cicatrización de una lesión; Sustitución, modificación o apoyo de la anatomía o de un proceso fisiológico; Soporte de vida; Control de la concepción; Desinfección de dispositivos médicos; Sustancias desinfectantes; Provisión de información mediante un examen in vitro de muestras extraídas del cuerpo humano, con fines diagnósticos; Dispositivos que incorporan tejidos de origen animal y/o humano, y/o Dispositivos empleados en fertilización in vitro y tecnologías de reproducción asistida; Y cuya finalidad de uso principal no es a través de mecanismos farmacológicos, inmunológicos o metabólicos, sin embargo, pueden ser asistidos por estos medios para lograr su función. Los dispositivos médicos incluyen a los insumos para la salud de las siguientes categorías: equipo médico, prótesis, órtesis, ayudas funcionales, agentes de diagnóstico, insumos de uso odontológico, materiales quirúrgicos, de curación y productos higiénicos. 7. Etapas del proceso de obtención del Registro Sanitario para dispositivos médicos Clase I, II, y III. Los trámites a verificar con apoyo de ésta guía, serán los incluidos en el ACUERDO por el que se modifica el diverso por el que se dan a conocer los trámites y servicios, así como los formatos que aplica la Secretaría de Salud, a través de la Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS), inscritos en el Registro Federal de Trámites y Servicios de la Comisión Federal de Mejora Regulatoria (CONAMER), publicado el 28 de enero de 2011 y el diverso por el cual se dan a conocer los formatos de los trámites a cargo de la Secretaría de Salud que se indican, publicado el 2 de septiembre de 2015, publicado en el Diario Oficial de la Federación el 24 de enero de 2022 (ACUERDO de trámites). Las etapas involucradas en la obtención del registro sanitario y por ende en la integración del dossier del dispositivo médico son las siguientes: 7.1. ¿El dispositivo médico requiere de registro sanitario? 7.2. Identificación de la categoría de uso del dispositivo médico 7.3. Clasificación de acuerdo con el riesgo sanitario. 7.4. Identificación de Homoclave y Modalidad 7.5. Requisitos legales, administrativos y técnicos para el trámite de Registro Sanitario El primer paso para comenzar con el proceso para la obtención del Registro Sanitario es tener la seguridad de que el dispositivo médico efectivamente requiere de registro sanitario, para lo cual es importante que el usuario consulte el “Acuerdo por el que se da a conocer el listado de insumos para la salud considerados como de bajo riesgo para efectos de obtención del Registro Sanitario, y de aquellos productos que por su naturaleza, características propias y uso no se consideran como insumos para la salud y por 17 ende no requieren Registro Sanitario, publicado en el Diario Oficial de la Federación. Para comenzar con la aplicación de los requisitos y por lo tanto con la integración de la información del dossier debemos identificar a que categoría pertenece el dispositivo médico a registrar. De acuerdo con el artículo 262 de la Ley General de Salud los grupos en los cuales se divide al sector de los dispositivos médicos en México con base en su función y finalidad de uso son los siguientes: I. Equipo médico. Los aparatos, accesorios e instrumental para uso específico, destinados a la atención médica, quirúrgica o a procedimientos de exploración, diagnóstico, tratamiento y rehabilitación de pacientes, así como aquellos para efectuar actividades de investigación biomédica. Nota: los equipos médicos requieren calibración, mantenimiento, reparación, desinstalación y disposición final, de igual manera es necesaria la capacitación del usuario. El equipo médico es utilizado para propósitos de monitoreo, diagnóstico y tratamiento de enfermedades o rehabilitación después de una enfermedad o lesión; estos pueden ser utilizados solos o en combinación con cualquier accesorio, consumible u otro equipo médico. El equipo médico excluye a los dispositivos médicos implantables, desechables o de un solo uso. Definiciones: Calibración 16 : Conjunto de operaciones que tiene como finalidad determinar los errores de un instrumento de medida respecto de sus valores nominales de funcionamiento. 14 Accesorio de un DM: Artículo que, sin ser en sí mismo un DM, está destinado por su fabricante a ser usado de forma conjunta con uno o varios de dichos productos, para permitir específicamente que el producto pueda utilizarse para su indicación de uso o para contribuir específica y directamente a la funcionalidad médica de los DM. Estos pueden venir incluidos dentro del mismo empaque del dispositivo médico o bien pueden suministrarse por separado como un accesorio para uso exclusivo con el dispositivo principal. 14 Consumibles : A los materiales necesarios para que el dispositivo médico realice sus funciones, desechables, que con su operación se agotan y que son de consumo repetitivo. II. Prótesis, órtesis y ayudas funcionales. Aquellos dispositivos destinados a sustituir o complementar una función, un órgano, o un tejido del cuerpo humano. III. Agentes de diagnóstico. Todos los insumos incluyendo antígenos, anticuerpos, calibradores, verificadores, reactivos, equipos de reactivos, medios de cultivo y de contraste y cualquier otro similar que pueda utilizarse como auxiliar de otros procedimientos clínicos o paraclínicos. Nota: Los agentes de diagnóstico son dispositivos médicos utilizados solos o en combinación con otros dispositivos médicos para proporcionar información para la detección, pronóstico, diagnóstico o monitoreo de condiciones fisiológicas, estados de salud, enfermedades o malformaciones congénitas en humanos. Nota: también se consideran los materiales y sustancias que se aplican a otros dispositivos médicos y áreas quirúrgicas con la finalidad de desinfectar, descontaminar y/o sanitizar. 8 VI. Productos higiénicos. Los materiales y substancias que se apliquen en la superficie de la piel o cavidades corporales y que tengan acción farmacológica o preventiva. Nota: la finalidad de uso de estos productos es ayudar a mantener la salud y/o prevenir la transmisión de enfermedades. VII. Los demás insumos que sean considerados para este uso y sean evaluados y reconocidos como dispositivos médicos por la Secretaría de Salud a solicitud. Existe una categoría reciente de dispositivos médicos la cual aún no ha sido autorizada en la Ley General de Salud vigente, sin embargo, ya aparece en el suplemento para dispositivos médicos de la Farmacopea de los Estados Unidos Mexicanos versión 5.0 como: VII. Software como dispositivo médico, al utilizado con uno o más propósitos médicos, que tiene como característica principal que no requiere formar parte del hardware del dispositivo médico para cumplir con el propósito médico previsto; es capaz de funcionar en plataformas computacionales generales y puede utilizarse solo y/o en combinación con otros productos (ej., como módulo, otros dispositivos médicos, etcétera). Las aplicaciones móviles que cumplen con esta definición son consideradas software como dispositivo médico. El software que hace funcionar al dispositivo médico físico queda excluido de esta definición. Una vez identificada la categoría de uso del Dispositivo Médico a registrar, se procede a la clasificación del mismo de acuerdo con el riesgo para la salud, la cual está sujeta a la indicación de uso y tiempo de permanencia o contacto con el cuerpo humano. 6 De acuerdo al artículo 83 del RIS, los dispositivos médicos se clasificarán para efectos de registro de acuerdo con el riesgo que implica su uso, de la manera siguiente: Clase I. Aquellos insumos conocidos en la práctica médica y que su seguridad y eficacia están comprobadas y, generalmente, no se introducen al organismo. Clase II. Aquellos insumos conocidos en la práctica médica y que pueden tener variaciones en el material con el que están elaborados o en su concentración y, generalmente, se introducen al organismo permaneciendo menos de treinta días. Clase III. Aquellos insumos nuevos o recientemente aceptados en la práctica médica, o bien que se introducen al organismo y permanecen en él, por más de treinta días. Nota: Para realizar una correcta clasificación del Dispositivo Médico a registrar se les sugiere revisar el Apéndice II. Normativo. Criterios para la Clasificación de Dispositivos Médicos con base en su nivel de riesgo sanitario, del Suplemento para Dispositivos Médicos 5.0, Tomo II, FEUM. Para ingresar la solicitud de Registro Sanitario a COFEPRIS, se debe elegir la homoclave y modalidad correspondiente al dispositivo médico a registrar, las cuales se enlistan a continuación: Homoclave COFEPRIS-04-001-A Solicitud de Registro Sanitario de Dispositivos Médicos. Modalidad A.- Productos de Fabricación Nacional. Homoclave COFEPRIS-04-001-B Solicitud de Registro Sanitario de Dispositivos Médicos. Modalidad B.- Productos de Importación (Fabricación Extranjera). Homoclave COFEPRIS-04-001-C Solicitud de Registro Sanitario de Dispositivos Médicos. Modalidad C.- Productos de Fabricación Nacional que son Maquilados por otro Establecimiento. Una vez identificada la categoría y la clasificación con base al riesgo sanitario se procede a la consulta de cada uno de los checklist correspondiente a Clase I, Clase II, Clase III y el correspondiente al Software como dispositivo médico, los cuales tienen como propósito servir como guía de requisitos que garantizarán la seguridad y eficacia del dispositivo médico. Nota: La información técnica, científica y clínica (evidencia que garantiza la seguridad y eficacia del dispositivo médico) que integrará el dossier, dependerá de la indicación de uso, descripción y clasificación de acuerdo con el riesgo. Checklist Dispositivos Médicos Clase I Checklist Dispositivos Médicos Clase II Checklist Dispositivos Médicos Clase III Checklist Software como Dispositivo Médico (ScDM) Clase I, ll y III 1. Constitución Política de los Estados Unidos Mexicanos. 2. Ley General de Salud. 3. Ley Federal de Procedimiento Administrativo. 4. Ley Federal de Derechos. 5. Reglamento de la Comisión Federal para la Protección contra Riesgos Sanitarios. 6. Reglamento de Insumos para la Salud. 7. Reglamento de la Ley Federal sobre Metrología y Normalización. 8. Reglamento de la Ley General de Salud en Materia de Publicidad. 9. Norma Oficial Mexicana NOM-001-SSA1-vigente, Que instituye la estructura de la Farmacopea de los Estados Unidos Mexicanos y sus suplementos y el procedimiento para su revisión, actualización, edición y difusión. 10. Norma Oficial Mexicana NOM-137-SSA1-vigente, Etiquetado de dispositivos médicos. 11. Acuerdo por el que se modifica el diverso por el que se dan a conocer los trámites y servicios, así como los formatos que aplica la Secretaría de Salud, a través de la Comisión Federal para la Protección contra Riesgos Sanitarios, inscritos en el Registro Federal de Trámites y Servicios de la Comisión Federal de Mejora Regulatoria, publicado el 28 de enero de 2011 y el diverso por el cual se dan a conocer los formatos de los trámites a cargo de la Secretaría de Salud que se indican, publicado el 2 de septiembre de 2015, publicado el 24 de enero de 2022. 12. Acuerdo por el que se modifica y adiciona el diverso por el que se dan a conocer los trámites y servicios, así como los formatos que aplica la Secretaría de Salud, a través de la Comisión Federal para la Protección contra Riesgos Sanitarios, inscritos en el Registro Federal de Trámites y Servicios de la Comisión Federal de Mejora Regulatoria, vigente. 13. Norma Oficial Mexicana NOM-241-SSA1-vigente, respecto a las buenas prácticas de fabricación de dispositivos médicos. 14. Farmacopea de los Estados Unidos Mexicanos. Suplemento para Dispositivos Médicos 5.0. 15. Guía para la Evaluación Clínica de Dispositivos Médicos. [Recurso electrónico]. México: Secretaría de Salud, Centro Nacional de Excelencia Tecnológica en Salud; 2017 16. Glosario de Gestión de Equipo Médico. México: Secretaría de Salud, Centro Nacional de Excelencia Tecnológica en Salud; 2016. 17. Acuerdo por el que se da a conocer el listado de insumos para la salud considerados como de bajo riesgo para efectos de obtención del Registro Sanitario, y de aquellos productos que por su naturaleza, características propias y uso no se consideran como insumos para la salud y por ende no requieren Registro Sanitario. www.dof.gob.mx/nota_detalle.php?codigo=5376857&fecha=22/12/2014#gsc.tab=0 www.dof.gob.mx/nota_detalle.php?codigo=5227732&fecha=31/12/2011#gsc.tab=0 12 13 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.1. Formato de solicitud. 1.2. Acreditación de representante legal. 1.3. Pago de derechos. FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 153 RIS Art. 14 LFPA Acuerdo de trámites y servicios Art. 15 LFPA Art. 195-A LFD Acuerdo de trámites y servicios. ELEMENTO QUE INTEGRA EL REQUISITO Original de solicitud Original u original de copia certificada del poder notarial o en su caso RUPA en copia simple y vigente. Original Comprobante pago CRITERIO Formato publicado por Cofepris para llevar a cabo la solicitud de trámites relacionados con el registro del producto. Formato vigente de autorizaciones, certificados y visitas. Debidamente llenado, los rubros que no apliquen a su solicitud deben estar cancelados con una línea paralela, firmada por el representante legal o responsable sanitario. Presentar original u original de copia certificada del Poder Notarial a favor del Representante Legal establecido en México mediante el que la empresa le confiere poderes amplios para realizar trámites de registros de los dispositivos médicos (artículo 262 LGS). Se podrá aceptar copia simple, debiendo hacer referencia al número de trámite en el cual ingreso el documento. El Representante Legal deberá ser el mismo que firme el formato de Solicitudes, o en su caso deberá ser firmada por el responsable sanitario. a. b. c. Cuota establecida por la LFD Para el caso de pago en banco: Sello bancario y fecha Impresión del pago electrónico Formatos presentados a la Autoridad para notificar los datos del establecimiento donde físicamente se lleva a cabo una actividad relacionada con el proceso de dispositivos médicos adicional al domicilio fiscal del propio establecimiento. Copia o número de aviso de funcionamiento con los siguientes elementos o características: 1.4. Avisos de funcionamiento y de Responsable Sanitario. Art. 181 RIS Art. 259 LGS NOM-241-SSA1-vigente Copia simple del aviso de funcionamiento Se presente en copia simple con los datos: a) Responsable Sanitario: b) Cédula Profesional: En el caso de solicitar incluir más de un distribuidor se deberá presentar los avisos de funcionamiento y de Responsable correspondientes -Razón social de la Empresa: - Domicilio: -Clasificación Autorizada: -Líneas de Fabricación: debe incluir la del producto solicitado -Fecha de Expedición -Firmado -Número de entrada que acredita su recepción en la COFEPRIS a. El aviso debe corresponder con los datos del establecimiento asentados en el formato. b. Incluir o anexar copia simple del aviso (s) c. Los datos expresados en la clave SCIAN deben corresponder al giro del establecimiento conforme a la actividad realizada. Con nombre y firma del propietario o representante legal. El responsable sanitario debe coincidir con la persona que avala la información técnica. El responsable sanitario deberá de cumplir con lo indicado en la norma oficial mexicana aplicable. 14 INFORMACIÓN ADMINISTRATIVA Y LEGAL FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Información completa o complementaria (contra etiqueta) en idioma español en los términos de la NOM 137 SSA1-vigente. Se pueden utilizar de manera opcional los símbolos incluidos en los apéndices normativos A e informativo B de dicha norma. 1.5. Proyectos de etiqueta o contra etiqueta. Art. 179 fracc. II RIS NOM 137-SSA1-vigente Proyecto de etiqueta o contraetiqueta en formato físico (copia impresa legible). Los dispositivos médicos estériles reusables deben indicar en el instructivo de uso o proyecto de etiqueta o contra etiqueta (marbete) la metodología a emplear para su re esterilización. La leyenda específica de productos estériles. Leyenda alusiva indicando que el producto es desechable o no reusable (cuando aplique). El proyecto de etiqueta se entrega en un tanto y en archivo de Word. Deberá incluir el listado de presentaciones con la descripción y número de código o catálogo de cada presentación del dispositivo médico (cuando aplique). Que deben corresponder a los señalados en certificado de libre venta y documentación técnica/comercial presentada. 1.6. Certificado de Libre Venta (CLV). Art. 180 fracc. I RIS Art. 153 RIS APÉNDICE III Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Certificado de Libre Venta o equivalente expedido por la autoridad sanitaria del país de origen. Documentos equivalentes: 1. Carta expedida por la autoridad sanitaria del país de origen donde se indique que dichos productos no están sujetos a control sanitario. 2. Documento emitido por el Ministerio correspondiente que regule los dispositivos médicos en el país de origen. 3. Reportes de los estudios clínicos concluyentes llevados a cabo en territorio nacional de acuerdo a lo señalado en el Reglamento de la Ley General de Salud en Materia de Investigación, con protocolo de investigación previamente autorizado por esta comisión. 4. Para el caso de que el CLV no avale al fabricante legal o para la autorización del fabricante legal sin CLV, deberán presentar documento legal notariado de origen que acredite la relación entre los fabricantes como responsabilidad legal para asegurar el cumplimiento de requerimientos regulatorios y responsabilidad en la calidad del producto a registrar. En este caso, presentar también el CLV del país de origen del fabricante del producto. Únicamente para fabricación extranjera: a. b. c. d. Documento legal autenticado. Original u original de copia certificada por notario en México. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses. Dicho documento deberá avalar: Producto (denominación distintiva y modelos), Fabricante Legal y/o Sitio de fabricación, lista de códigos de presentaciones. En caso de no incluir la lista de códigos de las presentaciones, deberá presentar carta aclaratoria emitida por el fabricante que avale dichos códigos. 15 INFORMACIÓN ADMINISTRATIVA Y LEGAL FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO Fabricación nacional: Oficio de Certificación de Buenas Prácticas de Fabricación emitido por la COFEPRIS. Fabricación extranjera: Certificado de Buenas Prácticas de Fabricación emitido por la autoridad sanitaria del país de origen o su documento equivalente. 1.7 Certificado de buenas prácticas de fabricación del (os) establecimiento (s) del fabricante (s) real (es) del (os) dispositivo médico o su documento equivalente. 1.8 Convenio de maquila (si aplica). Art. 179 fracc. VI RIS Art. 180 fracc. III RIS Art. 153 RIS Acuerdo de trámites y servicios. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Equivalentes: Certificado ISO 13485 versión vigente emitido por organismo autorizado. Certificado de marca CE para dispositivos médicos emitido por organismo autorizado en la Unión Europea. Declaración de cumplimiento de Buenas Prácticas de Fabricación incluida dentro del Certificado de Libre Venta emitido por la Autoridad Sanitaria o en su caso por el Ministerio correspondiente que regule el producto. CRITERIO Fabricación nacional: Oficio de Certificación de buenas prácticas de fabricación emitido por la COFEPRIS: 1. Copia simple y que exprese: razón social, domicilio, fecha de emisión y vigencia. 2. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. Fabricación extranjera: Original u original de copia certificada de Certificado de buenas prácticas de fabricación o documento equivalente del sitio o sitios de fabricación. 1. Documento legal autenticado. 2. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses 3. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. 4. El Certificado ISO 13485 puede ser autenticado en el país del fabricante real, legal o país de organismo certificador. Para fabricación nacional: Convenio de maquila firmado por ambas partes ante notario público. Para fabricación extranjera (fabricantes subcontratados): Documento legal que acredite la relación entre los fabricantes, responsabilidad legal de asegurar el cumplimiento de requerimientos regulatorios, sus responsabilidades en la calidad del producto a registrar, notariado de origen. (Solo se entregará la parte en donde se establece la relación de manufactura, responsabilidades en la calidad de productos y/o suministro). Copia simple del documento, para ambos casos. 16 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.9 Carta de representación (solo cuando el producto no es fabricado por la casa matriz, subsidiaria o filial del solicitante del registro en México) FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO Carta de representación original u original de copia certificada. Art. 16 RIS Art. 180 fracc. II RIS Art. 161 fracc. III RIS Acuerdo de trámites y servicios. En caso de solicitar incluir más de un distribuidor se deberá presentar la carta de representación para cada uno de ellos. y/o en su caso original de copia certificada de Carta de representación o aclaratoria en la cual un distribuidor tiene la facultad a su vez de nombrar a otros como sus distribuidores. CRITERIO Documento emitido por el fabricante legal responsable en la cual otorga los derechos de comercialización y distribución de sus productos a una compañía establecida en territorio nacional. a. Solo debe presentarse si el producto no es fabricado por la casa matriz o filial de la compañía, fábrica o laboratorio que solicite el registro en México. b. En Original u original de copia certificada en original, en idioma español o traducido al español por perito traductor. c. Documento legal autenticado. (7) d. Emitido por el fabricante legal en el extranjero. e. Otorgado a la compañía que solicita el registro en México y en su caso a otros distribuidores e importadores. En caso de ser filial, deberá presentar carta de filiales. 1.10 Declaración de aval de responsable sanitario ante la COFEPRIS Art. 153 RIS Carta original de declaración de aval de responsable en donde avala la información técnica presentada debidamente firmada por el responsable sanitario así como de las traducciones simples de la información técnica en caso de presentarse en idioma diferente al español o inglés. 17 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.1 Información general 2.2. Instructivo, si procede, para su uso, inserto o manual de operación APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Art. 179 fracc. III RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. NOM 137 SSA1-vigente Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO Documento emitido por el fabricante o solicitante del registro en México (en idioma español). Instructivo, inserto o manual de operación en formato físico (copia impresa legible) en idioma español. CRITERIO Información General: 1. Denominación genérica. 2. Denominación distintiva. 3. Descripción de las características y especificaciones técnicas 4. Forma física o farmacéutica y cuando aplique composición o fórmula cuali-cuantitativa 5. Presentaciones. En el caso de contar con varias presentaciones, se debe incluir el listado de presentaciones del producto que incluya códigos (claves) y la descripción en su caso, pudiendo incluir el catálogo comercial que las contenga sólo con fines de Información. 6. Indicación de uso. 7. La categoría y clasificación con base al nivel de riesgo sanitario, entre otros. 8. Listado de accesorios para el registro de equipos, se debe incluir el listado de accesorios que se suministren con el mismo para que sean incluidos en el oficio deregistro, cuando aplique. Deberá contener como mínimo la siguiente información dependiendo del tipo de producto: 1. Denominación distintiva y en su caso denominación genérica (conforme al Art. 2, fracción IV del RIS). 2. Descripción. 3. Finalidad de uso. 4. Presentaciones. 5. Listado de componentes o partes (cuando aplique). 6. Ensamble y desensamble (cuando aplique). 7. Operación, limpieza y esterilización (cuando aplique) 8. Condiciones de conservación y almacenamiento (cuando aplique.) 9. Mantenimiento (cuando aplique). 10. Calibración (cuando aplique). 11. Precauciones. 12. Preparación (cuando aplique). 13. Advertencias y leyendas alusivas correspondientes (cuando aplique). 14. Contraindicaciones (cuando aplique). 15. Incidentes adversos (cuando aplique). 16. Para productos formulados, se debe incluir la vía de administración, forma farmacéutica y contenido de ingrediente activo. 17. Técnica quirúrgica (cuando aplique). 18 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.3 Descripción o diagrama de las partes funcionales del producto 2.4 Declaración de la fórmula cuali- cuantitativa por unidad de medida, dosis o porcentual para productos formulados emitida por el fabricante Art. 179 RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 179 RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitariode un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS. ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Descripción, imagen, composición o diagrama de las partes funcionales. Aplica para dispositivos médicos no formulados y sus accesorios a. Señalar de qué partes está conformado el producto (equipos médicos principalmente). b. Lista de materiales de elaboración utilizados en el dispositivo médico indicando el nombre y composición de los materiales que se integran o incluyen en el mismo, señalando la función que desempeñan. c. Diagrama o esquema de diseño con especificaciones dimensionales que represente el dispositivo médico. Declaración de fórmula cuali-cuantitativa por unidad de medida, dosis o porcentual, para productos formulados, incluyendo todos los ingredientes del producto, debidamente firmada por el responsable de la calidad del fabricante en el país de origen, avalada por el responsable sanitario del establecimiento que solicita el registro sanitario en México. Deberá declarar: Documento que contenga la descripción de la formula cuali- cuantitativa (con su traducción al idioma español). Ingredientes Activos - Nombre químico, nombre genérico o descripción común internacional (DCI) y nombre comercial del ingrediente activo cuando proceda. Aditivos. La información relativa a los aditivos debe incluir (si es aplicable): - Descripción y contenido de cada uno de ellos, estén o no en el producto final. 2.5 Materias primas Art. 179 RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Copia de los Certificados de análisis de las materias primas con los cuales se demuestre identidad, pureza, esterilidad, cuando proceda, inocuidad, seguridad, estabilidad, según aplique. Los Certificados de análisis de materia prima deberán corresponder a aquellas materias primas que estarán en contacto con tejidos o fluidos y con soluciones para administración, con resultados del análisis químico, pruebas físicas, mecánicas y demás establecidas de acuerdo a FEUM, Farmacopeas internacionales o estándares internacionales, emitidos por los fabricantes o proveedores y firmados por el responsable de calidad. Para productos formulados, presentar certificado de análisis de los componentes de la fórmula. Certificado de análisis de materia prima (acero inoxidable, titanio y demás aplicables) para dispositivos médicos no formulados que están en contacto con el paciente o fluidos corporales, los cuales deberán mostrar cumplimiento con especificaciones conforme a estándares internacionales o monografías farmacopeícas. 19 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Pruebas microbiológicas, de funcionalidad conforme a las normas vigentes. y desempeño En caso de reactivos analíticos que contengan hemoderivados (componentes de origen humano) deberán de presentar el certificado de ausencia de anticuerpos de VIH y hepatitis como mínimo. Reportes completos de las pruebas eléctricas, pudiendo ser de manera enunciativa más no limitativa Reporte CB, Reporte UL, IECEE standard, ISO standard etc., según aplique. Cuando aplique de acuerdo a las características del dispositivo médico Pruebas de laboratorio especificaciones del insumo. 2.6 Pruebas de laboratorio Art. 179 fracc. VII RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS para verificar las En caso de utilizar metodología que no sea farmacopeica o que cumpla con un estándar internacional, deberá de presentar la descripción de los métodos de análisis o metodología (procedimientos) y su resumen del informe de validación del método que emplea el fabricante para verificar el cumplimiento de especificaciones, de acuerdo a la naturaleza y características del dispositivo, excepto equipo médico. Reportes de pruebas realizadas para comprobar que la naturaleza y el tiempo de contacto de un producto satisfaga las especificaciones de seguridad establecidas ya sea conforme: 1. La FEUM, u otras farmacopeas reconocidas o Normas Oficiales Mexicanas o pruebas desarrolladas conforme a especificaciones de organismos especializados (incluir referencia del estándar internacional correspondiente), u otra bibliografía científica reconocida internacionalmente. 2. Las pruebas de biocompatibilidad pueden llevarse a cabo ya sea en producto terminado o en las materias primas utilizadas para la elaboración del producto. 3. Para materiales ampliamente conocidos se podrán utilizar los resúmenes de pruebas aplicados a otros dispositivos médicos del mismo material y uso pretendido, siempre que se justifique mediante la información técnica con la que se pueda correlacionar a un producto equivalente el cual muestra similitud en composición y finalidad de uso, deberá estar basado conforme al proceso de gestión de riesgos con base a la ISO 10993-1. 4. Para todos los materiales ampliamente reconocidos y que su calidad y seguridad biológica está sustentada por una norma internacional (ISO, ASTM, ADA, DIN, ANSI), se podrán presentar las referencias bibliográficas del material con el certificado de análisis que avale la materia prima. (Todo insumo que este en contacto con el paciente, con fluidos corporales y soluciones suministradas al paciente deberán presentar los reportes de biocompatibilidad completos sin excepción). 20 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.7 Información del proceso de fabricación 2.8 Esterilización para aquellos productos que ostenten la leyenda de estéril 2.9 Información de envase ELEMENTO QUE INTEGRA EL REQUISITO Se puede presentar a través de: Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Descripción o diagrama de flujo del proceso de fabricación del producto. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Información sobre el proceso de esterilización. Protocolo de la validación del proceso de esterilización, reporte y certificado de esterilidad completo. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un Dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS CRITERIO Descripción de los elementos del envase primario y en su caso secundario a. Diagrama de flujo del proceso de manufactura que identifique cada uno de los pasos, subproceso y controles en proceso, emitido por el o los fabricantes, o b. Descripción general del proceso de fabricación que identifique cada uno de los pasos, subproceso y controles en proceso. Debe ser específico para el dispositivo o familia de dispositivos a registrar. Emitido por el fabricante. Cuando aplique deberán presentar: 1. Tipo de proceso de esterilización. 2. Resumen de la validación del proceso de esterilización del producto en el envase primario propuesto con breve descripción del proceso que se llevó a cabo con los parámetros evaluados, resultados obtenidos y conclusiones; que incluya como mínimo la calificación física y desempeño microbiológico con el que se garantice un SAL 10-6 realizado conforme al estándar internacional aplicable. 3. Certificado de esterilidad con resultados microbiológicos, emitido y avalado por el responsable de la evaluación (solo en caso de que en el certificado analítico de producto terminado no se contemple la esterilidad del producto). 4. En el caso de que la esterilización sea por óxido de etileno se deben de presentar los resultados de la prueba de residuos de óxido de etileno. 5. En caso de que el dispositivo médico estéril fuera reusable, se deben presentar los reportes y documentación que demuestre que el producto mantiene sus especificaciones originales durante los procesos de re esterilización indicados por el fabricante. 5.1. Los dispositivos médicos reusables o que se tengan que esterilizar previo a su uso deben indicar en el instructivo de uso o proyecto de etiqueta o contra etiqueta (marbete) la metodología a emplear para su re esterilización y el número de ciclos que pueda aplicarse al producto. 6. Los dispositivos médicos estériles desechables deben incluir en el proyecto de etiqueta o contra etiqueta (marbete) una leyenda alusiva indicando que el producto es desechable o no reusable. Descripción del envase primario y en su caso el secundario, señalando los materiales con el nombre químico y especificaciones dimensionales en la fabricación del mismo, emitido por el fabricante. 21 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Documentos equivalentes al certificado de análisis, que pueden presentarse de acuerdo al tipo de dispositivo, presentados de manera enunciativa mas no limitativa: 1. Documento emitido por el responsable de la calidad de la fabricación del producto en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie. En este caso deberá presentarse por separado los criterios de aceptación o especificaciones avalados por el responsable de la calidad del producto. 2.10 Certificado analítico o certificado de producto terminado. Art. 179 RIS Art. 180 fracc. IV RIS NOM-241- SSA1-vigente APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Documento emitido por el responsable de la calidad del producto o por el fabricante en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie, deberá estar emitido en hoja con membrete de la razón social del responsable de su emisión. 2. Declaración de Conformidad emitida por el fabricante o por el responsable de la calidad de la fabricación u organismo autorizado en donde se señale que el producto cumple con los requisitos establecidos para su comercialización y uso con base a normas internacionales de calidad, anexando los reportes completos de pruebas correspondientes solo en el caso de que sea específico para un número de lote o de serie del dispositivo a registrar. Así mismo, cuando la declaración no lo indique, deberá presentarse por separado el documento complementario que avalen la seguridad y eficacia tales como: Los parámetros evaluados y su criterio de aceptación firmado por el responsable de calidad del fabricante Estudios IEC, o Audit Report, o Product Performance Qualification (PPQ), o Master Validation Report (MVR), o Design Verification Report (DVR) 3. Copia del resumen del reporte de verificación del dispositivo médico (“Device Verification Report” / “Test Verification Report”). Se podrá presentar en original o copia simple, con firma autógrafa o electrónica, de conformidad con las buenas prácticas de fabricación y con el sistema de calidad del fabricante, según aplique. 22 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA 2.11 Estudios de estabilidad para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 85 RIS NOM-241- SSA1-vigente Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS APÉNDICE IX. Normativo. Estudios de estabilidad para Dispositivos médicos, Suplemento para dispositivos médicos 5.0, FEUM. ELEMENTO QUE INTEGRA EL REQUISITO Reportes con conclusiones de los estudios de estabilidad en el envase(s) primario(s) propuesto(s), que avalen el periodo de caducidad. Pruebas de integridad del empaque para productos estériles que estén en contacto directo con el paciente y fluidos corporales. CRITERIO Estudios de estabilidad. Para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad, se deberá evaluar que el dispositivo conserva sus propiedades físicas, químicas y biológicas, características de calidad establecidas, para cumplir con el uso para el cual fue diseñado. Se debe presentar el resumen el estudio de estabilidad (acelerado o en tiempo real) el cual contenga: Nombre del dispositivo médico, presentaciones No. de lotes evaluados, tamaño del lote, Composición del empaque (cuando aplique), Condiciones del estudio, Parámetros de prueba, criterios de aceptación y referencia de los métodos de análisis, Tiempos de muestreo y análisis, con resultados analíticos por condición de almacenamiento y fechas de análisis, Evaluación de datos y conclusiones. Dicho resumen y sus conclusiones deberá estar revisado y firmado por el responsable de la unidad de calidad del fabricante. Los estudios de estabilidad pueden ser acelerados (siempre y cuando sean concluyentes y contengan el resumen del protocolo, reportes de resultados y conclusiones) y deberá presentar el reporte del avance de estabilidad en tiempo real en caso de contar con ellos. Para confirmar el plazo de caducidad, deberá presentar los resultados de los reportes del estudio de estabilidad en tiempo real una vez concluidos. El fabricante deberá establecer la metodología y condiciones de prueba basado en las características del producto que garanticen que durante el periodo de vida útil recomendado se conservan las características originales. Para dispositivos cuya naturaleza lo requiera (como reactivos de diagnóstico, soluciones oftálmicas, soluciones para conservación/limpieza/desinfección de lentes de contacto) deberán incluir reporte de las pruebas de estabilidad en uso para establecer el periodo de tiempo de uso y la consideración de almacenamiento, ambos indicados en su etiqueta, durante el cual un dispositivo para su uso una vez reconstituido o diluido o multidosis, , puede ser usado mientras cumpla con las especificaciones, una vez abierto el envase primario y haya sido utilizado por primera vez. 23 24 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.1. Formato de solicitud. 1.2. Acreditación de representante legal. FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 153 RIS Art. 14 LFPA. Acuerdo de trámites y servicios. Art. 15 LFPA ELEMENTO QUE INTEGRA EL REQUISITO Original de solicitud Original u original de copia certificada del poder notarial o en su caso RUPA copia simple y vigente. CRITERIO Formato publicado por Cofepris para llevar a cabo la solicitud de trámites relacionados con el registro del producto. Formato vigente de autorizaciones, certificados y visitas. Debidamente llenado y firmado por el propietario, representante legal o responsable sanitario. Presentar original u original de copia certificada del Poder Notarial a favor del Representante Legal establecido en México mediante el que la empresa le confiere poderes amplios para realizar trámites de registros de los dispositivos médicos (artículo 262 LGS). Se podrá aceptar copia simple, debiendo hacer referencia al número de trámite en el cual ingreso el documento. El Representante Legal deberá ser el mismo que firme el formato de Solicitudes, o en su caso deberá ser firmada por el responsable sanitario. 1.3. Pago de derechos Art. 195-A LFD. Acuerdo de trámites y servicios. 1.4. Avisos de funcionamiento y de Responsable Sanitario. Art. 181 RIS Art. 259 LGS NOM-241-SSA1-vigente Original Comprobante pago Copia simple del aviso de funcionamiento Se presente en copia simple con los datos: a) Responsable Sanitario: b) Cédula Profesional: En el caso de solicitar incluir más de un distribuidor se deberá presentar los avisos de funcionamiento y de Responsable correspondientes a. Cuota establecida por la LFD b. Para el caso de pago en banco: Sello bancario y fecha c. Impresión del pago electrónico Formatos presentados a la Autoridad para notificar los datos del establecimiento donde físicamente se lleva a cabo una actividad relacionada con el proceso de dispositivos médicos adicional al domicilio fiscal del propio establecimiento. Copia o número de aviso de funcionamiento con los siguientes elementos o características: -Razón social de la Empresa: -Domicilio: -Clasificación Autorizada: -Líneas de Fabricación: debe incluir la del producto solicitado -Fecha de Expedición -Firmado -Número de entrada que acredita su recepción en la COFEPRIS a. El aviso debe corresponder con los datos del establecimiento asentados en el formato. b. Incluir o anexar copia simple del aviso (s) c. Los datos expresados en la clave SCIAN deben corresponder al giro del establecimiento conforme a la actividad realizada. Con nombre y firma del propietario o representante legal. El responsable sanitario debe coincidir con la persona que avala la información técnica. El responsable sanitario deberá de cumplir con lo indicado en la norma oficial mexicana aplicable. 25 INFORMACIÓN ADMINISTRATIVA Y LEGAL FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 1.5 Proyectos de etiqueta o contra etiqueta 1.6. Certificado de Libre Venta (CLV). Art. 179 fracc. II RIS. NOM 137-SSA1-vigente. Art 180 fracc. I RIS Art. 153 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Proyecto de etiqueta o contraetiqueta en formato físico (copia impresa legible). Información completa o complementaria (contra etiqueta) en idioma español en los términos de la NOM 137 SSA1-vigente. Se pueden utilizar de manera opcional los símbolos incluidos en los apéndices normativos A e informativo B de dicha norma. Los dispositivos médicos estériles reusables deben indicar en el instructivo de uso o proyecto de etiqueta o contra etiqueta (marbete) la metodología a emplear para su re esterilización. La leyenda específica de productos estériles. Leyenda alusiva indicando que el producto es desechable o no reusable (cuando aplique). El proyecto de etiqueta se entrega en un tanto y en archivo de Word. Deberá incluir el listado de presentaciones con la descripción y número de código o catálogo de cada presentación del dispositivo médico (cuando aplique). Que deben corresponder a los señalados en certificado de libre venta y documentación técnica/comercial presentada. Certificado de Libre Venta o equivalente expedido por la autoridad sanitaria del país de origen. Documentos equivalentes: 1. Carta expedida por la autoridad sanitaria del país de origen donde se indique que dichos productos no están sujetos a control sanitario. 2. Documento emitido por el Ministerio correspondiente que regule los dispositivos médicos en el país de origen. 3. Reportes de los estudios clínicos concluyentes llevados a cabo en territorio nacional de acuerdo a lo señalado en el Reglamento de la Ley General de Salud en Materia de Investigación, con protocolo de investigación previamente autorizado por esta comisión. 4. Para el caso de que el CLV no avale al fabricante legal o para la autorización del fabricante legal sin CLV, deberán presentar documento legal notariado de origen que acredite la relación entre los fabricantes como responsabilidad legal para asegurar el cumplimiento de requerimientos regulatorios y responsabilidad en la calidad del producto a registrar. En este caso, presentar también el CLV del país de origen del fabricante del producto. Únicamente para fabricación extranjera: a. Documento legal autenticado. (7) b. Original u original de Copia certificada por notario en México. c. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses. d. Dicho documento deberá avalar: Producto (denominación distintiva y modelos), Fabricante Legal y/o Sitio de fabricación, lista de códigos de presentaciones. En caso de no incluir la lista de códigos de las presentaciones, deberá presentar carta aclaratoria emitida por el fabricante que avale dichos códigos. 26 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.7 Certificado de buenas prácticas de fabricación del (os) establecimiento (s) del fabricante (s) real (es) del (os) dispositivo médico o su documento equivalente. 1.8 Convenio de maquila (si aplica). FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 179 fracc .VI RIS Art. 180 fracc. III RIS Art. 153 RIS Acuerdo de trámites y servicios. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. ELEMENTO QUE INTEGRA EL REQUISITO Fabricación nacional: Oficio de Certificación de Buenas Prácticas de Fabricación emitido por la COFEPRIS. Fabricación extranjera: Certificado de Buenas Prácticas de Fabricación emitido por la autoridad sanitaria del país de origen o su documento equivalente. Equivalentes: Certificado ISO 13485 versión vigente emitido por organismo autorizado. Certificado de marca CE para dispositivos médicos emitido por organismo autorizado en la Unión Europea. Declaración de cumplimiento de Buenas Prácticas de Fabricación incluida dentro del Certificado de Libre Venta emitido por la Autoridad Sanitaria o en su caso por el Ministerio correspondiente que regule el producto. Para fabricación nacional: Convenio de maquila firmado por ambas partes ante notario público. Para fabricación extranjera (fabricantes subcontratados): Documento legal que acredite la relación entre los fabricantes, responsabilidad legal de asegurar el cumplimiento de requerimientos regulatorios, sus responsabilidades en la calidad del producto a registrar, notariado de origen. (Solo se entregará la parte en donde se establece la relación demanufactura, responsabilidades en la calidad de productos y/o suministro). CRITERIO Fabricación nacional: Oficio de Certificación de buenas prácticas de fabricación emitido por la COFEPRIS: 1. Copia simple y que exprese: razón social, domicilio, fecha de emisión y vigencia. 2. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. Fabricación extranjera: Original u original de copia certificada de Certificado de buenas prácticas de fabricación o documento equivalente del sitio o sitios de fabricación. 1. Documento legal autenticado. 2. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses. 3. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. 4. El Certificado ISO 13485 puede ser autenticado en el país del fabricante real, legal o país de organismo certificador. Copia simple del documento, para ambos casos. 27 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.9 Carta de representación (solo cuando el producto no es fabricado por la casa matriz, subsidiaria o filial del solicitante del registro en México). 1.10 Declaración de aval de responsable sanitario ante la COFEPRIS. FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO Carta de representación original u original de copia certificada. Art. 16 RIS Art. 180 fracc. II RIS Art. 161 fracc. III RIS Acuerdo de trámites y servicios. Art. 153 RIS En caso de solicitar incluir más de un distribuidor se deberá presentar la carta de representación para cada uno de ellos y/o en su caso original de copia certificada de Carta de representación o aclaratoria en la cual un distribuidor tiene la facultad a su vez de nombrar a otros como sus distribuidores. CRITERIO Documento emitido por el fabricante legal responsable en la cual otorga los derechos de comercialización y distribución de sus productos a una compañía establecida en territorio nacional. a. Solo debe presentarse si el producto no es fabricado por la casa matriz o filial de la compañía, fábrica o laboratorio que solicite el registro en México. b. En original u original copia certificada en original, en idioma español o traducido al español por perito traductor. c. Documento legal autenticado. d. Emitido por el fabricante legal en el extranjero. e. Otorgado a la compañía que solicita el registro en México y en su caso a otros distribuidores e importadores. En caso de ser filial, deberá presentar carta de filiales. Carta original de declaración de aval de responsable en donde avala la información técnica presentada debidamente firmada por el responsable sanitario así como de las traducciones simples de la información técnica en caso de presentarse en idioma diferente al español o inglés. 28 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.1. Información general 2.2 Instructivo, si procede, para su uso, inserto o manual de operación APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Art. 179 fracc. III RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. NOM-137-SSA1-vigente Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO Documento emitido por el fabricante o solicitante del registro en México (en idioma español). Instructivo, inserto o manual de operación en formato físico (copia impresa legible) en idioma español. CRITERIO Información General: 1. Denominación genérica. 2. Denominación distintiva. 3. Descripción de las características y especificaciones técnicas 4. Forma física o farmacéutica y cuando aplique composición o fórmula cuali-cuantitativa 5. Presentaciones. En el caso de contar con varias presentaciones, se debe incluir el listado de presentaciones del producto que incluya códigos (claves) y la descripción en su caso, pudiendo incluir el catálogo comercial que las contenga sólo con fines de Información. 6. Indicación de uso. 7. La categoría y clasificación con base al nivel de riesgo sanitario, entre otros. 8. Listado de accesorios para el registro de equipos, se debe incluir el listado de accesorios que se suministren con el mismo para que sean incluidos en el oficio de registro, cuando aplique. Deberá contener como mínimo la siguiente información dependiendo del tipo de producto: 1. Denominación distintiva y en su caso denominación genérica (conforme al Art. 2, fracción IV del RIS). 2. Descripción. 3. Finalidad de uso. 4. Presentaciones 5. Listado de componentes o partes (cuando aplique)6. Ensamble y desensamble (cuando aplique) 7. Operación, limpieza y esterilización (cuando aplique). 8. Condiciones de conservación y almacenamiento (cuando aplique) 9. Mantenimiento (cuando aplique)10. Calibración (cuando aplique). 11. Precauciones. 12. Preparación (cuando aplique). 13. Advertencias y leyendas alusivas correspondientes (cuando aplique). 14. Contraindicaciones (cuando aplique). 15. Incidentes adversos (cuando aplique). 16. Para medios de contraste y productos formulados, se debe incluir la vía de administración, forma farmacéutica y contenido de ingrediente activo. 17. Técnica quirúrgica (cuando aplique). 29 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.3 Descripción o diagrama de las partes funcionales del producto. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. ELEMENTO QUE INTEGRA EL REQUISITO Descripción, imagen, composición o diagrama de las partes funcionales. CRITERIO Aplica para dispositivos médicos no formulados y sus accesorios a. Señalar de qué partes está conformado el producto (equipos médicos principalmente). b. Lista de materiales de elaboración utilizados en el dispositivo médico indicando el nombre y composición de los materiales que se integran o incluyen en el mismo, señalando la función que desempeñan. c. Diagrama o esquema de diseño con especificaciones dimensionales que represente el dispositivo médico. (Aplica para dispositivos médicos que estén en contacto con el paciente, con tejido o fluidos corporales así como con soluciones destinadas a administrarse al paciente). Declaración de fórmula cuali-cuantitativa por unidad de medida, dosis o porcentual, para productos formulados, incluyendo todos los ingredientes del producto, debidamente firmada por el responsable de la calidad del fabricante en el país de origen, avalada por el responsable sanitario del establecimiento que solicita el registro sanitario en México. 2.4 Declaración de la fórmula cuali-cuantitativa por unidad de medida, dosis o porcentual para productos formulados emitida por el fabricante. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Documento que contenga la descripción de la formula cuali- cuantitativa (con su traducción al idioma español). Deberá declarar: Ingredientes Activos -Nombre químico, nombre genérico o descripción común internacional (DCI) y nombre comercial del ingrediente activo cuando proceda. – Aditivos. La información relativa a los aditivos debe incluir (si es aplicable): -Descripción y contenido de cada uno de ellos, estén o no en el producto final. 2.5 Materias primas Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Copia de los Certificados de análisis de las materias primas con los cuales se demuestre identidad, pureza, esterilidad, cuando proceda, inocuidad, seguridad, estabilidad, según aplique. Los Certificados de análisis de materia prima deberán corresponder a aquellas materias primas que estarán en contacto con tejidos o fluidos y con soluciones para administración, con resultados del análisis químico, pruebas físicas, mecánicas y demás establecidas de acuerdo a FEUM, Farmacopeas internacionales o estándares internacionales, emitidos por los fabricantes o proveedores y firmados por el responsable de calidad. Para productos formulados, presentar certificado de análisis de los componentes de la fórmula. Certificado de análisis de materia prima (acero inoxidable, titanio y demás aplicables) para dispositivos médicos no formulados que están en contacto con el paciente o fluidos corporales, los cuales deberán mostrar cumplimiento con especificaciones conforme a estándares internacionales o monografías farmacopeícas. 30 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO Pruebas de laboratorio para verificar las especificaciones del insumo. 2.6 Pruebas de laboratorio Art. 179 fracc. VII RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS En caso de utilizar metodología que no sea farmacopeica o que cumpla con un estándar internacional, deberá de presentar la descripción de los métodos de análisis o metodología (procedimientos) y su resumen del informe de validación del método que emplea el fabricante para verificar el cumplimiento de especificaciones, de acuerdo a la naturaleza y características del dispositivo, excepto equipo médico. CRITERIO De acuerdo a las características, naturaleza y finalidad de uso del dispositivo, se podrá presentar: a)Reportes completos de las pruebas realizadas por el fabricante para validar y/o verificar el diseño, funcionalidad y/o desempeño del dispositivo y demostrar que satisface las especificaciones establecidas ya sea conforme a la FEUM, u otras farmacopeas reconocidas o Normas Oficiales Mexicanas o pruebas desarrolladas conforme a especificaciones de organismos especializados (incluir referencia del estándar internacional correspondiente), u otra bibliografía científica reconocida internacionalmente. También podrán ser pruebas desarrolladas por el fabricante cuando no exista referencia en la FEUM ni se encuentren comprendidos en referencias o normas internacionales (“Device Verification Report” / “Test Verification Report”). Dichos reportes deberán incluir procedimientos, métodos de prueba o descripción de la metodología, identificación de la muestra evaluada, parámetros evaluados, criterios de aceptación y resultados, firmados por el responsable de la evaluación. Reportes de estudios aplicables a sueros hemoclasificadores y condones, fabricados por laboratorios nacionales y extranjeros se deben presentar los resultados de evaluación (eficacia) emitidos por un laboratorio autorizado por la Secretaría de Salud. De no existir un laboratorio autorizado con el panel necesario para efectuar la prueba, deberá demostrarse adjuntando la carta emitida por dicho laboratorio informando de esta situación, y en este caso se podrán presentar resultados de evaluación efectuados en el extranjero. b) Reportes completos de las pruebas eléctricas, pudiendo ser de manera enunciativa más no limitativa Reporte CB, Reporte UL, IECEE standard, ISO standard etc., según aplique. Se puede Incluir carta aclaratoria siempre que sea pertinente en virtud de que las pruebas fueron realizadas en otro dispositivo (tipo comparativo) con misma indicación de uso y mismo materiales de elaboración. En el caso de equipo médico se incluirán las pruebas específicas conforme IEC60601 de seguridad eléctrica, compatibilidad electromagnética y de funcionalidad específicas eléctricas que le aplican a ese producto. Además de la validación del software cuando aplique. En el caso de insumos que incluyan otro dispositivo médico como coadyuvante para su función o envase primario, por ejemplo: en donde el envase primario es un dispositivo médico (ejemplo jeringa), catéteres de entrega para otros dispositivos, agujas para administración del implante o para suturas, estos deben cumplir con todas las pruebas de funcionalidad, seguridad y eficacia conforme a la legislación vigente aplicable. En caso de productos hemoderivados que contengan componentes de origen humano deberán de presentar el certificado de ausencia de anticuerpos de VIH y hepatitis como mínimo. 31 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Se puede presentar a través de: 2.7 Información del proceso de fabricación. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Descripción o diagrama de flujo del proceso de fabricación del producto. a. Diagrama de flujo del proceso de manufactura que identifique cada uno de los pasos, subproceso y controles en proceso, emitido por el o los fabricantes, o b. Descripción general del proceso de fabricación que identifique cada uno de los pasos, subproceso y controles en proceso. Debe ser específico para el dispositivo o familia de dispositivos a registrar. Emitido por el fabricante. Cuando aplique deberán presentar: 2.8 Esterilización para aquellos productos que ostenten la leyenda de estéril. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Información sobre el proceso de esterilización. Protocolo de la validación del proceso de esterilización, reporte y certificado de esterilidad completo. 1. Tipo de proceso de esterilización. 2. Resumen de la validación del proceso de esterilización del producto en el envase primario propuesto con breve descripción del proceso que se llevó a cabo con los parámetros evaluados, resultados obtenidos y conclusiones; que incluya como mínimo la calificación física y desempeño microbiológico con el que se garantice un SAL 10-6 realizado conforme al estándar internacional aplicable. 3. Certificado de esterilidad con resultados microbiológicos, emitido y avalado por el responsable de la evaluación (solo en caso de que en el certificado analítico de producto terminado no se contemple la esterilidad del producto.) 4. En el caso de que la esterilización sea por óxido de etileno se deben de presentar los resultados de la prueba de residuos de óxido de etileno. 5. En caso de que el dispositivo médico estéril fuera reusable, se deben presentar los reportes y documentación que demuestre que el producto mantiene sus especificaciones originales durante los procesos de re esterilización indicados por el fabricante. 5.1. Los dispositivos médicos reusables o que se tengan que esterilizar previo a su uso deben indicar en el instructivo de uso o proyecto de etiqueta o contra etiqueta (marbete) la metodología a emplear para su re esterilización y el número de ciclos que pueda aplicarse al producto. 6. Los dispositivos médicos estériles desechables deben incluir en el proyecto de etiqueta o contra etiqueta (marbete) una leyenda alusiva indicando que el producto es desechable o no reusable. 32 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA 2.9 Información de envase FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO Descripción de los elementos del envase primario y en su caso secundario CRITERIO Descripción del envase primario y en su caso el secundario, señalando los materiales con el nombre químico y especificaciones dimensionales en la fabricación del mismo, emitido por el fabricante. Documentos equivalentes al certificado de análisis, que pueden presentarse de acuerdo al tipo de dispositivo, presentados de manera enunciativa mas no limitativa: 2.10 Certificado analítico o certificado de producto terminado. Art. 179 RIS Art. 180 fracc. IV RIS NOM-241-SSA1-vigente APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Documento emitido por el responsable de la calidad del producto o por el fabricante en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie, deberá estar emitido en hoja con membrete de la razón social del responsable de su emisión. 1. Documento emitido por el responsable de la calidad de la fabricación del producto en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie. En este caso deberá presentarse por separado los criterios de aceptación o especificaciones avalados por el responsable de la calidad del producto. 2. Declaración de Conformidad emitida por el fabricante o por el responsable de la calidad de la fabricación u organismo autorizado en donde se señale que el producto cumple con los requisitos establecidos para su comercialización y uso con base a normas internacionales de calidad, anexando los reportes completos de pruebas correspondientes solo en el caso de que sea específico para un número de lote o de serie del dispositivo a registrar. Así mismo, cuando la declaración no lo indique, deberá presentarse por separado el documento complementario que avalen la seguridad y eficacia tales como: Los parámetros evaluados y su criterio de aceptación firmado por el responsable de calidad del fabricante Estudios IEC, o Audit Report, o Product Performance Qualification (PPQ), o Master Validation Report (MVR), o Design Verification Report (DVR) 3. Copia del resumen del reporte de verificación del dispositivo médico (“Device Verification Report” / “Test Verification Report”). Se podrá presentar en original o copia simple, con firma autógrafa o electrónica, de conformidad con las buenas prácticas de fabricación y con el sistema de calidad del fabricante, según aplique. 33 2.11 Estudios de estabilidad para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad Art. 85 RIS NOM-241- SSA1-vigente Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS APÉNDICE IX. Normativo. Estudios de estabilidad para Dispositivos médicos, Suplemento para dispositivos médicos 5.0, FEUM. Reportes con conclusiones de los estudios de estabilidad en el envase(s) primario(s) propuesto(s), que avalen el periodo de caducidad. Pruebas de integridad del empaque para productos estériles que estén en contacto directo con el paciente y fluidos corporales. Estudios de estabilidad. Para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad, se deberá evaluar que el dispositivo conserva sus propiedades físicas, químicas y biológicas, características de calidad establecidas, para cumplir con el uso para el cual fue diseñado. Se debe presentar el resumen el estudio de estabilidad (acelerado o en tiempo real) el cual contenga: Nombre del dispositivo médico, presentaciones No. de lotes evaluados, tamaño del lote, Composición del empaque (cuando aplique), Condiciones del estudio, Parámetros de prueba, criterios de aceptación y referencia de los métodos de análisis, Tiempos de muestreo y análisis, con resultados analíticos por condición de almacenamiento y fechas de análisis, Evaluación de datos y conclusiones. Dicho resumen y sus conclusiones deberá estar revisado y firmado por el responsable de la unidad de calidad del fabricante. Los estudios de estabilidad pueden ser acelerados (siempre y cuando sean concluyentes y contengan el resumen del protocolo, reportes de resultados y conclusiones) y deberá presentar el reporte del avance de estabilidad en tiempo real en caso de contar con ellos. Para confirmar el plazo de caducidad, deberá presentar los resultados de los reportes del estudio de estabilidad en tiempo real una vez concluidos. El fabricante deberá establecer la metodología y condiciones de prueba basado en las características del producto que garanticen que durante el periodo de vida útil recomendado se conservan las características originales. Para dispositivos cuya naturaleza lo requiera (como reactivos de diagnóstico, soluciones oftálmicas, soluciones para conservación/limpieza/desinfección de lentes de contacto) deberán incluir reporte de las pruebas de estabilidad en uso para establecer el periodo de tiempo de uso y la consideración de almacenamiento, ambos indicados en su etiqueta, durante el cual un dispositivo para su uso una vez reconstituido o diluido o multidosis, , puede ser usado mientras cumpla con las especificaciones, una vez abierto el envase primario y haya sido utilizado por primera vez. 34 FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Reportes de pruebas realizadas para comprobar que la naturaleza y el tiempo de contacto de un producto satisfaga las especificaciones de seguridad establecidas ya sea conforme: 2.12 Pruebas de biocompatibilidad. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. MGA-DM 10993-1. Pruebas de biocompatibilidad. Evaluación y pruebas dentro de un proceso de gestión de riesgos, Suplemento para Dispositivos Médicos 5.0 Reportes completos de las pruebas de biocompatibilidad aplicables según el tipo de dispositivo 1. La FEUM, u otras farmacopeas reconocidas o Normas Oficiales Mexicanas o pruebas desarrolladas conforme a especificaciones de organismos especializados (incluir referencia del estándar internacional correspondiente), u otra bibliografía científica reconocida internacionalmente. 2. Las pruebas de biocompatibilidad pueden llevarse a cabo ya sea en producto terminado o en las materias primas utilizadas para la elaboración del producto. 3. Para materiales ampliamente reconocidos se podrán utilizar los resúmenes de pruebas aplicados a otros dispositivos médicos del mismo material y uso pretendido, siempre que se justifique mediante la información técnica con la que se pueda correlacionar a un producto equivalente el cual muestra similitud en composición y finalidad de uso, deberá estar basado conforme al proceso de gestión de riesgos con base a la ISO 10993-1. 4. Para todos los materiales ampliamente reconocidos y que su calidad y seguridad biológica está sustentada por una norma internacional (ISO, ASTM, ADA, DIN, ANSI), se podrán presentar las referencias bibliográficas del material con el certificado de análisis que avale la materia prima. (Todo insumo que este en contacto con el paciente, con fluidos corporales y soluciones suministradas al paciente deberán presentar los reportes de biocompatibilidad completos sin excepción). 35 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Copia de las publicaciones en revistas indexadas de los estudios clínicos concluyentes y con la suficiente casuística, realizados para demostrar la seguridad y eficacia del dispositivo a registrar. 2.13 Estudios clínicos (cuando aplique). APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Evaluación clínica que contenga el análisis exhaustivo de los datos clínicos disponibles, relativos a la finalidad de uso del dispositivo a registrar, incluyendo datos sobre su desempeño y seguridad. Deberá incluir datos específicos sobre el dispositivo a registrar, así como cualquier dato relacionado con los dispositivos que el fabricante considere comparables con el suyo. Ambos dispositivos deben tener la misma finalidad de uso y deberán ser comparados con respecto a sus características técnicas y biológicas. La evaluación deberá ser realizada por profesionales expertos en el área. Para dispositivos nuevos que no cuenten con estudios clínicos en otros países, presentar el reporte y conclusiones de estudios clínicos realizados en México, con previa autorización del protocolo de investigación, los cuales deberán ser concluyentes y con la suficiente casuística, anexar copia de las publicaciones correspondientes. Traducción completa del reporte en caso de presentarse en idioma diferente al español y/o inglés. En caso de las publicaciones, deberán presentar 3 como mínimo, siempre que sea concluyente y con la muestra poblacional suficiente para demostrar la seguridad y eficacia. Sí no hay publicaciones específicas para el dispositivo a registrar, se puede considerar una evaluación realizada por profesionales expertos en el área, mediante la cual sustenten la similitud o equivalencia con productos que ya demostraron su seguridad y eficacia, debiendo tratase de los mismos materiales, tecnología e indicación de uso del dispositivo médico a registrar. Adicional presentar las publicaciones más representativas de los Dispositivos Médicos con los que es comparable (mínimo una referencia). 36 37 FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.1. Formato de solicitud 1.2. Acreditación de representante legal 1.3. Pago de derechos 1.4. Avisos de funcionamiento y de Responsable Sanitario Art.153 RIS Art. 14 LFPA Acuerdo de trámites y servicios. Art. 15 LFPA Art. 195-A LFD Acuerdo de trámites y servicios. Art. 181 RIS Art. 259 LGS NOM-241-SSA1vigente ELEMENTO QUE INTEGRA EL REQUISITO Original de solicitud. Original u original de copia certificada del poder notarial, o en su caso RUPA copia simple y vigente. Original Comprobante pago Copia simple del aviso de funcionamiento Se presente en copia simple con los datos: a) Responsable Sanitario b) Cédula Profesional En el caso de solicitar incluir más de un distribuidor se deberá presentar los avisos de funcionamiento y de Responsable correspondientes. CRITERIO Formato publicado por Cofepris para llevar a cabo la solicitud de trámites relacionados con el registro del producto. Formato vigente de autorizaciones, certificados y visitas. Debidamente llenado y firmado por el propietario, representante legal o responsable sanitario. Presentar original u original de copia certificada del Poder Notarial a favor del Representante Legal establecido en México mediante el que la empresa le confiere poderes amplios para realizar trámites de registros de los dispositivos médicos (artículo 262 LGS). Se podrá aceptar copia simple, debiendo hacer referencia al número de trámite en el cual ingreso el documento. El Representante Legal deberá ser el mismo que firme el formato de Solicitudes, o en su caso deberá ser firmada por el responsable sanitario. a. b. c. Cuota establecida por la LFD Para el caso de pago en banco: Sello bancario y fecha Impresión del pago electrónico Formatos presentados a la Autoridad para notificar los datos del establecimiento donde físicamente se lleva a cabo una actividad relacionada con el proceso de dispositivos médicos adicional al domicilio fiscal del propio establecimiento. Copia o número de aviso de funcionamiento con los siguientes elementos o características: -Razón social de la Empresa: -Domicilio: -Clasificación Autorizada: -Líneas de Fabricación: debe incluir la del producto solicitado -Fecha de Expedición -Firmado -Número de entrada que acredita su recepción en la COFEPRIS a. El aviso debe corresponder con los datos del establecimiento asentados en el formato. b. Incluir o anexar copia simple del aviso (s) c. Los datos expresados en la clave SCIAN deben corresponder al giro del establecimiento conforme a la actividad realizada. -Con nombre y firma del propietario o representante legal. El responsable sanitario debe coincidir con la persona que avala la información técnica. El responsable sanitario deberá de cumplir con lo indicado en la norma oficial mexicana aplicable. 38 FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.5. Proyectos de etiqueta o contra etiqueta Art. 179 fracc. II RIS NOM 137-SSA1vigente ELEMENTO QUE INTEGRA EL REQUISITO Proyecto de etiqueta o contraetiqueta en formato físico (copia impresa legible). CRITERIO Información completa o complementaria (contra etiqueta) en idioma español en los términos de la NOM 137 SSA1-vigente. Se pueden utilizar de manera opcional los símbolos incluidos en los apéndices normativos A e informativo B de dicha norma. Los dispositivos médicos estériles reusables deben indicar en el instructivo de uso o proyecto de etiqueta o contra etiqueta (marbete) la metodología a emplear para su re esterilización. El proyecto de etiqueta se entrega en un tanto y en archivo de Word. Deberá incluir el listado de presentaciones con la descripción y número de código o catálogo de cada presentación del dispositivo médico (cuando aplique), que deben corresponder a los señalados en certificado de libre venta y documentación técnica/comercial presentada. Certificado de Libre Venta o equivalente expedido por la autoridad sanitaria del país de origen. 1.6. Certificado de Libre Venta (CLV). Art 180 fracc. I RIS Art. 153 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Documentos equivalentes: 1. Carta expedida por la autoridad sanitaria del país de origen donde se indique que dichos productos no están sujetos a control sanitario. 2. Documento emitido por el Ministerio correspondiente que regule los dispositivos médicos en el país de origen. 3. Reportes de los estudios clínicos concluyentes llevados a cabo en territorio nacional de acuerdo a lo señalado en el Reglamento de la Ley General de Salud en Materia de Investigación, con protocolo de investigación previamente autorizado por esta comisión. 4. Para el caso de que el CLV no avale al fabricante legal o para la autorización del fabricante legal sin CLV, deberán presentar documento legal notariado de origen que acredite la relación entre los fabricantes como responsabilidad legal para asegurar el cumplimiento de requerimientos regulatorios y responsabilidad en la calidad del producto a registrar. En este caso, presentar también el CLV del país de origen del fabricante del producto. Únicamente para fabricación extranjera: a. Documento legal autenticado (7) b. Original u original de copia certificada por notario en México. c. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses. d. Dicho documento deberá avalar: Producto (denominación distintiva y modelos), Fabricante Legal y/o Sitio de fabricación, lista de códigos de presentaciones. En caso de no incluir la lista de códigos de las presentaciones, deberá presentar carta aclaratoria emitida por el fabricante que avale dichos códigos. 39 INFORMACIÓN ADMINISTRATIVA Y LEGAL FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO Fabricación nacional: Oficio de Certificación de Buenas Prácticas de Fabricación emitido por la Cofepris. 1.7 Certificado de buenas prácticas de fabricación del (os) establecimiento (s) del fabricante (s) real (es) del (os) dispositivo médico o su documento equivalente. Fabricación extranjera: Certificado de Buenas Prácticas de Fabricación emitido por la autoridad sanitaria del país de origen o su documento equivalente Art. 179 fracc. VI RIS Art. 180 fracc. III RIS Art. 153 RIS Equivalentes: •Certificado ISO 13485 versión vigente emitido por organismo autorizado. • Certificado de marca CE para dispositivos médicos emitido por organismo autorizado en la Unión Europea. •Declaración de cumplimiento de Buenas Prácticas de Fabricación incluida dentro del Certificado de Libre Venta emitido por la Autoridad Sanitaria o en su caso por el Ministerio correspondiente que regule el producto. CRITERIO Fabricación nacional: Certificado de buenas prácticas de fabricación emitido por la COFEPRIS: 1. Copia simple y que exprese: razón social, domicilio, fecha de emisión y vigencia. 2. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. Fabricación nacional de radiofármacos o que maneje radiación ionizante: 1. Copia de la Licencia de operación para radiofarmacia y Autorización para adquisición o transferencia, emitidos por la Comisión Nacional de Seguridad Nuclear y Salvaguardias. Fabricación extranjera: Original u original de copia certificada de Certificado de buenas prácticas de fabricación o documento equivalente del sitio o sitios de fabricación. 1. Documento legal autenticado (7) 2. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses 3. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. 4. El Certificado ISO 13485 puede ser autenticado en el país del fabricante o país de organismo certificador. 1.8 Convenio de maquila en caso de proceder Acuerdo de trámites y servicios. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Para fabricación nacional: Convenio o contrato de maquila firmado por ambas partes ante notario público. Para fabricación extranjera (fabricantes subcontratados): Documento legal que acredite la relación entre los fabricantes, responsabilidad legal de asegurar el cumplimiento de requerimientos regulatorios, sus responsabilidades en la calidad del producto a registrar notariado de origen. (Solo se entregará la parte en donde se establece la relación de manufactura, responsabilidades en la calidad de productos y/o suministro). Copia simple del documento, para ambos casos. 40 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.9 Carta de representación (solo cuando el producto no es fabricado por la casa matriz, subsidiaria o filial del solicitante del registro en México). FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 16 RIS Art. 180 fracc. II RIS Art. 161 fracc. III RIS Acuerdo de trámites y servicios. ELEMENTO QUE INTEGRA EL REQUISITO Carta de representación original u original de copia certificada. En caso de solicitar incluir más de un distribuidor se deberá presentar la carta de representación para cada uno de ellos. O en su caso original de copia certificada de Carta de representación o aclaratoria en la cual un distribuidor tiene la facultad a su vez de nombrar a otros como sus distribuidores. CRITERIO Documento emitido por el fabricante legal responsable en la cual otorga los derechos de comercialización y distribución de sus productos a una compañía establecida en territorio nacional. a. Solo debe presentarse si el producto no es fabricado por la casa matriz o filial de la compañía, fábrica o laboratorio que solicite el registro en México. b. En original u original de copia certificada en original, en idioma español o traducido al español por perito traductor. c. Documento legal autenticado. (7) d. Emitido por el fabricante legal en el extranjero. e. Otorgado a la compañía que solicita el registro en México y en su caso a otros distribuidores e importadores. En caso de ser filial, deberá presentar carta de filiales. 1.10 Declaración de aval de responsable sanitario ante la COFEPRIS Carta original de declaración de aval de responsable en donde avala la información técnica presentada debidamente firmada por el responsable sanitario. Art. 153 RIS Y de las traducciones simples de la información técnica en caso de presentarse en idioma diferente al español o inglés. 41 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.1 Información general 2.2 Instructivo, si procede, para su uso, inserto o manual de operación APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Art. 179 fracc. III RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. NOM 137-SSA1-vigente Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO Documento emitido por el solicitante del registro en México (en idioma español). Instructivo, inserto o manual de operación en formato físico (copia impresa legible) en idioma español. CRITERIO Información General: 1. Denominación genérica. 2. Denominación distintiva. 3. Descripción de las características y especificaciones técnicas 4. Forma física o farmacéutica y cuando aplique composición o fórmula cuali-cuantitativa. 5. Presentaciones. En el caso de contar con varias presentaciones, se debe incluir el listado de presentaciones del producto que incluya códigos (claves) y la descripción en su caso, pudiendo incluir el catálogo comercial que las contenga sólo con fines de Información. 6. Indicación de uso. 7. La categoría y clasificación con base al nivel de riesgo sanitario, entre otros. 8. Listado de accesorios, cuando aplique, se debe incluir el listado de accesorios que se suministren con el mismo para que sean incluidos en el oficio de registro, cuando aplique. Deberá contener como mínimo la siguiente información dependiendo del tipo de producto: 1. Denominación distintiva y en su caso denominación genérica (conforme al Art. 2, fracción IV del RIS). 2. Descripción. 3. Finalidad de uso. 4. Presentaciones. 5. Listado de componentes o partes (cuando aplique). 6. Ensamble y desensamble (cuando aplique). 7. Operación, limpieza y esterilización (cuando aplique). 8. Condiciones de conservación y almacenamiento (cuando aplique). 9. Mantenimiento (cuando aplique.) 10. Calibración (cuando aplique). 11. Precauciones 12. Preparación (cuando aplique). 13. Advertencias y leyendas alusivas correspondientes (cuando aplique). 14. Contraindicaciones (cuando aplique). 15. Incidentes adversos (cuando aplique). 16. Para medios de contraste y productos formulados, se debe incluir la vía de administración, forma farmacéutica y contenido de ingrediente activo. 17. Técnica quirúrgica (cuando aplique). 42 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.3 Descripción o diagrama de las partes funcionales del producto. 2.4 Declaración de la fórmula cualicuantitativa por unidad de medida, dosis o porcentual para productos formulados emitida por el fabricante. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 153 RIS Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Descripción, imagen, composición o diagrama de las partes funcionales. Aplica para dispositivos médicos no formulados y sus accesorios. a. Señalar de qué partes y componentes está conformado el producto (dispositivos activos). b. Lista de materiales de elaboración utilizados en el dispositivo médico indicando el nombre y composición de los materiales (de los que están en contacto con tejido, mucosas, sangre) que se integran o incluyen en el mismo (Para dispositivos activos). c. Lista de materiales de elaboración utilizados en el dispositivo médico indicando el nombre químico y composición de los materiales debiendo declarar colorantes, material radiopaco, tintas, etc., según aplique, señalando la función que desempeñan y los que tienen contacto con tejidos, sangre, mucosas. (Materiales quirúrgicos, de curación, prótesis, ayudas funcionales). d. Dibujo o imagen esquemática que represente el dispositivo médico (Equipo médico o dispositivo activo). e. Para otros dispositivos, cuando aplique, esquemas de diseño o dibujos de ingeniería emitidos por el fabricante, con especificaciones dimensionales que representen al dispositivo médico. Documento que contenga la descripción de la formula cuali- cuantitativa (con su traducción al idioma español). Declaración de fórmula cuali-cuantitativa por unidad de medida, dosis o porcentual, para productos formulados, incluyendo todos los ingredientes del producto, debidamente firmada por el responsable de la calidad del fabricante en el país de origen, avalada por el responsable sanitario del establecimiento que solicita el registro sanitario en México. Deberá declarar: Ingredientes Activos - Nombre químico, nombre genérico o descripción común internacional (DCI) y nombre comercial del ingrediente activo cuando proceda. Aditivos. La información relativa a los aditivos debe incluir (si es aplicable): - Descripción y contenido de cada uno de ellos, estén o no en el producto final. 43 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Los Certificados de análisis deberán corresponder a aquellas materias primas que estarán en contacto con tejidos o fluidos, con resultados del análisis químico, pruebas físicas, mecánicas y demás establecidas de acuerdo a FEUM, Farmacopeas internacionales o estándares internacionales, emitidos por los fabricantes o proveedores y firmados por el responsable de calidad. Certificado de análisis de materia prima (acero inoxidable, titanio, aleaciones metálicas, polímeros y demás aplicables) para dispositivos médicos no formulados que están en contacto con el paciente o fluidos corporales, los cuales deberán mostrar cumplimiento con especificaciones conforme a estándares internacionales o monografías farmacopeicas. 2.5 Materias primas Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Para dispositivos que contienen algún fármaco como medios de contraste, agentes de diagnóstico que se administran directamente al paciente (in vivo), agentes de diagnóstico con isótopos radiactivos e inyectables así como dispositivos implantables, el solicitante de registro debe proporcionar la siguiente información relacionada con Copia de los Certificados de análisis de las materias los ingredientes activos y aditivos empleados en la fabricación del primas con los cuales se demuestre identidad, producto: Ingredientes Activos. pureza, esterilidad, cuando proceda, inocuidad, a. Nombre químico, nombre genérico o descripción común seguridad, estabilidad, según aplique. internacional (DCI) y nombre comercial del ingrediente activo cuando proceda. b. Estructura química, cuando proceda c. Características físicas y químicas. d. Información de la fabricación del (fármaco) ingrediente activo, cuando proceda. e. Nombre y domicilio del fabricante del fármaco con Certificado de Buenas Prácticas de Fabricación emitido por la autoridad Sanitaria del país de origen. f. Control del fármaco (ingrediente activo). La información que debe proporcionarse es la siguiente: Certificado de análisis del fármaco (ingrediente activo) con resultados que verifiquen el cumplimiento de especificaciones físicas, químicas, biológicas conforme a farmacopea, de ser el caso. Especificaciones del fármaco (ingrediente activo), incluyendo perfil de impurezas y disolventes residuales. Descripción de los métodos analíticos utilizados para la evaluación fármaco, indicando en su caso la monografía farmacopéica. Resumen de la validación con conclusiones, para el caso de métodos no farmacopéicos. 44 Justificación de las especificaciones cuando no sean farmacopéicas. Aditivos. La información relativa a los aditivos de los dispositivos médicos incluidos en el numeral de materias primas debe contener si es aplicable: a. Descripción de cada uno de ellos, se encuentren o no en el producto final. b. Copia del certificado de análisis del fabricante de cada uno de los aditivos, firmados por el responsable de la calidad, en su caso. Para productos derivados de tejido humano deben presentar la Licencia sanitaria o autorización emitida por la autoridad correspondiente del banco de tejidos del cual proceden los tejidos donados. Pruebas de laboratorio especificaciones del insumo. 2.6 Pruebas de laboratorio Art. 179 fracc. VII RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS para verificar las En caso de utilizar metodología que no sea farmacopeica o que cumpla con un estándar internacional, deberá de presentar la descripción de los métodos de análisis o metodología (procedimientos) que emplea el fabricante para verificar el cumplimiento de especificaciones, de acuerdo a la naturaleza y características del dispositivo. Para métodos de análisis fisicoquímicos y/o biológicos utilizados en dispositivos formulados o dispositivos que contengan un fármaco (implantables, medios de contraste, agentes de diagnóstico), en caso de no tratarse de métodos farmacopeícos deberá presentar el resumen del informe de validación de los mismos, con el cual se demuestre que es consistente y reproducible. De acuerdo a las características, naturaleza y finalidad de uso del dispositivo, se podrá presentar: a) Reportes completos de las pruebas realizadas por el fabricante para validar y/o verificar el diseño, funcionalidad y/o desempeño del dispositivo y demostrar que satisface las especificaciones establecidas ya sea conforme a la FEUM, u otras farmacopeas reconocidas o Normas Oficiales Mexicanas o pruebas desarrolladas conforme a especificaciones de organismos especializados (incluir referencia del estándar internacional correspondiente), u otra bibliografía científica reconocida internacionalmente. También podrán ser pruebas desarrolladas por el fabricante cuando no exista referencia en la FEUM ni se encuentren comprendidos en referencias o normas internacionales. (“DeviceVerificationReport” / “Test VerificationReport”) Dichos reportes deberán incluir los procedimientos / métodos de prueba o descripción de la metodología, identificación de la muestra evaluada, parámetros evaluados, criterios de aceptación y resultados, estar firmados por el responsable de la evaluación. b) Reportes completos de las pruebas eléctricas, pudiendo ser de manera enunciativa más no limitativa Reporte CB, Reporte UL, IECEE standard, ISO standard etc., según aplique. En caso de que las pruebas sean realizadas en un modelo diferente al dispositivo a registrar deberán presentar justificación técnica emitida por el fabricante responsable donde se demuestre que son comparables en materiales de elaboración, tecnología e intención de uso entre ambos dispositivos y que es aplicable al modelo o dispositivo a registrar. Se puede incluir carta aclaratoria siempre que sea pertinente en virtud de que las pruebas fueron realizadas en otro dispositivo (tipo comparativo) con la misma indicación de uso y materiales de elaboración. En el caso de equipo médico se incluirán las pruebas conforme IEC 60601 de seguridad eléctrica, compatibilidad electromagnética y de funcionalidad específicas que le aplique a ese dispositivo médico., además de la validación del software cuando aplique. 45 En el caso de insumos que incluyan otro dispositivo médico como coadyuvante para su función o envase primario, por ejemplo: en donde el envase primario es un dispositivo médico (ejemplo jeringa), catéteres de entrega para otros dispositivos, agujas para administración del implante o suturas, estos deben cumplir con todas las pruebas de funcionalidad, seguridad y eficacia conforme a la legislación vigente aplicable. En caso de reactivos analíticos que contengan hemoderivados (componentes de origen humano) deberán incluir el certificado de ausencia de anticuerpos de VIH y hepatitis como mínimo. Para el caso de dispositivos que contengan tejidos de origen animal, deberán incluir el certificado de origen de que el tejido es obtenido de un proveedor certificado así como que se encuentra libre de cualquier agente infecto contagioso (TSE, BSE, fiebre aftosa, leucosis bovina y otros que representen un riesgo a la salud.). Para dispositivos que contengan tejido o células de origen humano deberán incluir la documentación que demuestre la trazabilidad del origen del tejido (donante) hasta el producto obtenido como dispositivo médico, así como los reportes de las pruebas serológicas y pruebas para garantizar que está libre de ácidos nucleicos (mediante NAT) de los Virus de Inmunodeficiencia Humana (VIH) 1 y 2, Virus de Hepatitis A (VHA), Virus de Hepatitis B (VHB), Virus de Hepatitis C (VHC) o cualquier otro microorganismo infecto contagioso. Reportes de estudios aplicables a agentes de diagnóstico para determinación de hepatitis C, para antígeno de superficie de hepatitis B, sueros hemoclasificadores y VIH, fabricados por laboratorios nacionales y extranjeros se deben presentar los resultados de evaluación (eficacia) emitidos por un laboratorio autorizado por la Secretaría de Salud. De no existir un laboratorio autorizado con el panel necesario para efectuar la prueba, deberá demostrarse adjuntando la carta emitida por dicho laboratorio informando de esta situación, y en este caso se podrán presentar resultados de evaluación efectuados en el extranjero. 46 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO 2.7 Información del proceso de fabricación Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO Descripción o diagrama de flujo del proceso de fabricación del producto. CRITERIO Se puede presentar a través de: a. Diagrama de flujo del proceso de manufactura que identifique cada uno de los pasos, subprocesos y controles en proceso, así como etapas críticas, emitidos por el o los fabricantes, o b. Descripción general del proceso de fabricación que identifique cada uno de los pasos, subproceso y controles en proceso. Debe ser específico para el dispositivo o familia de dispositivos a registrar. Emitido por el fabricante. 2.8 Esterilización para aquellos productos que ostenten la leyenda de estéril Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Información sobre el proceso de esterilización. Protocolo de la validación del proceso de esterilización, reporte y certificado de esterilidad completo. Cuando aplique deberán presentar: 1. Tipo de proceso de esterilización. 2. Reporte de Validación del proceso de esterilización con breve descripción del proceso con los parámetros evaluados de acuerdo al proceso de esterilización utilizado, resultados obtenidos de calificación de desempeño físico y microbiológico y conclusiones que demuestren se obtiene un SAL mínimo de 10-6. Realizado conforme a estándar internacional aplicable. 3. Certificado de esterilidad y endotoxinas (cuando aplique) con resultados microbiológicos, emitido y avalado por el responsable de la evaluación (solo en caso de que en el certificado analítico de producto terminado no se contemple la esterilidad del producto.) 4. En el caso de que la esterilización sea por óxido de etileno se deben de presentar los resultados de la prueba de residuos de óxido de etileno. 5. Para medios de contraste y agentes de diagnóstico inyectables adicionalmente deben presentar información sobre la adecuabilidad del proceso de esterilización. 6. En caso de que el dispositivo médico estéril fuera reusable, se deben presentar los reportes y/o documentación que demuestre que el producto mantiene sus especificaciones originales durante los procesos de re esterilización indicados por el fabricante. 7. Los dispositivos médicos estériles desechables deben incluir en el proyecto de etiqueta o contra etiqueta (marbete) una leyenda alusiva indicando que el producto es desechable, de un solo uso o no reusable. 47 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Información sobre el envase. Para todos los dispositivos médicos excepto equipo médico el solicitante del registro debe incluir la siguiente información: 2.9 Información del envase Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS Descripción de los elementos del envase primario y en su caso secundario 1. Descripción del sistema de empaque (envase primario, secundario o sistema de barrera estéril), señalando los materiales usados en la fabricación del mismo con nombre químico o denominación común internacional, además del nombre comercial. 2. Pruebas de integridad del empaque para medios de contraste inyectables, agentes de diagnóstico inyectables, así como cualquier otro dispositivo que se suministre estéril con las cuales se garantice que el producto conserva sus características de calidad, estabilidad, hermeticidad y esterilidad en su caso, en los procesos de transporte y almacenamiento. 3. Para el caso de medios de contraste se deben presentar certificados de análisis emitidos por el fabricante del producto para cada uno de los materiales del sistema contenedor-cierre empleados. Documentos equivalentes al certificado de análisis, que pueden presentarse de acuerdo al tipo de dispositivo, presentados de manera enunciativa mas no limitativa: 2.10 Certificado analítico o certificado de producto terminado. Art. 179 RIS Art. 180 fracc. IV RIS NOM-241- SSA1-vigente APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Documento emitido por el responsable de la calidad del producto o por el fabricante en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie, deberá estar emitido en hoja con membrete de la razón social del responsable de su emisión. 1. Documento emitido por el responsable de la calidad de la fabricación del producto en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie. En este caso deberá presentarse por separado los criterios de aceptación o especificaciones avalados por el responsable de la calidad del producto. 2. Reportes de control de calidad (resultados de las pruebas realizadas durante el proceso de fabricación como al producto terminado), para verificar el cumplimiento de especificaciones, debiendo señalar especificaciones, resultados, número de lote o número de serie, firmado por el responsable de calidad. 3. Declaración de Conformidad emitida por el fabricante o por el responsable de la calidad de la fabricación u organismo autorizado en donde se señale que el producto cumple con los requisitos establecidos para su comercialización y uso con base a normas internacionales de calidad, anexando los reportes completos de pruebas correspondientes, solo en el caso de que sea específico para un número de lote o de serie del dispositivo a registrar. Así mismo, cuando la declaración no lo indique, deberá presentarse por separado el documento complementario que avale la seguridad y eficacia tales como: 48 Los parámetros evaluados y su criterio de aceptación firmado por el responsable de calidad del fabricante Estudios IEC, o Audit Report, o Product Performance Qualification (PPQ), o Master Validation Report (MVR), o Design Verification Report (DVR) 4. Copia del resumen del reporte de verificación del dispositivo médico (“Device Verification Report” / “Test Verification Report”). Consideraciones generales: Se podrá presentar en original o copia simple, con firma autógrafa o electrónica, de conformidad con las buenas prácticas de fabricación y con el sistema de calidad del fabricante, según aplique 2.11 Estudios de estabilidad para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad Art. 85 RIS NOM-241-SSA1-vigente Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. APÉNDICE IX. Normativo. Estudios de estabilidad para Dispositivos médicos, Suplemento para Dispositivos Médicos 5.0, FEUM. Art. 16 RIS Reportes completos con conclusiones de los estudios de estabilidad en el envase(s) primario(s) propuesto(s), que avalen el periodo de caducidad. Pruebas de integridad del empaque para productos estériles que estén en contacto directo con el paciente y fluidos corporales. Estudios de estabilidad. Para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad, se deberá evaluar que el dispositivo conserva sus propiedades físicas, químicas y biológicas, características de calidad establecidas, para cumplir con el uso para el cual fue diseñado. El reporte del estudio de estabilidad debe contener como mínimo: 1. Nombre del dispositivo médico y presentaciones 2. No. de lotes evaluados y tamaño del lote 3. Descripción del sistema de empaque (envase primario, sistema de barrera estéril y empaque secundario cuando éste sea una condición esencial para su vida útil) 4. Condiciones del estudio (Temperatura y humedad, ecuación del modelo matemático en caso de tratarse de estabilidad acelerada) 5. Parámetros de prueba, criterios de aceptación y referencia de los métodos de análisis 6. Tiempos de muestreo y análisis, con resultados analíticos por condición de almacenamiento y fechas de análisis 7. Evaluación de datos y conclusiones. a) b) c) d) Dicho reporte y sus conclusiones deberá estar revisado y firmado por el responsable de la unidad de calidad del fabricante El fabricante deberá establecer la metodología y condiciones de prueba basado en las características del producto que garanticen que durante el periodo de vida útil recomendado se conservan las características originales. Cuando se lleven a cabo estudios de estabilidad o envejecimiento acelerado, deben demostrar y/o justificar el modelo matemático utilizado. Los estudios se deben llevar a cabo con muestras representativas del proceso de producción en el envase o empaque primario propuesto para su almacenamiento y distribución bajo las condiciones establecidas por el fabricante, en un mínimo de tres lotes. 49 e) f) g) 2.12 Pruebas de biocompatibilidad Art. 179 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. MGA-DM 10993-1. Pruebas de biocompatibilidad. Evaluación y pruebas dentro de un proceso de gestión de riesgos, Suplemento para Dispositivos Médicos 5.0 Reportes completos de las pruebas de biocompatibilidad según el tipo del dispositivo, basado en un análisis de riesgos La vida útil o fecha de caducidad tentativa debe ser confirmada con estudios de estabilidad o envejecimiento en tiempo real (a largo plazo). Los estudios de estabilidad para dispositivos médicos formulados que contengan un fármaco deben realizarse conforme a los numerales aplicables de la Norma Oficial Mexicana NOM-073-SSA1-2015, Estabilidad de fármacos y medicamentos, así como de remedios herbolarios. Para dispositivos cuya naturaleza lo requiera (como reactivos de diagnóstico, soluciones oftálmicas, soluciones para conservación/limpieza/desinfección de lentes de contacto) deberán incluir reporte de las pruebas de estabilidad en uso para establecer el periodo de tiempo de uso y la consideración de almacenamiento, ambos indicados en su etiqueta, durante el cual un dispositivo para su uso una vez reconstituido o diluido o multidosis, , puede ser usado mientras cumpla con las especificaciones, una vez abierto el envase primario y haya sido utilizado por primera vez. Pruebas realizadas para comprobar que la naturaleza y el tiempo de contacto de un producto satisfaga las especificaciones de seguridad conforme a la FEUM, u otras farmacopeas reconocidas o Normas Oficiales Mexicanas o pruebas desarrolladas conforme a especificaciones de organismos especializados (incluir referencia del estándar internacional correspondiente), u otra bibliografía científica reconocida internacionalmente. Consideraciones: 1. Para materiales ampliamente conocidos se podrán utilizar los reportes de pruebas aplicados a otros dispositivos médicos del mismo material y uso pretendido, siempre que se justifique mediante la información técnica con la que se pueda correlacionar a un producto equivalente el cual muestra similitud en composición, características técnicas, biológicas y finalidad de uso. 2.Cuando la información presentada corresponda a la versión anterior del producto o a un producto similar se podrán presentar los reportes en función de los análisis de materiales empleados y no sobre el producto terminado. 3.Para todos los materiales ampliamente reconocidos y que su calidad y seguridad biológica está sustentada por una norma internacional (ISO, ASTM, ADA, DIN, ANSI), se podrán presentar las referencias bibliográficas del material con el certificado de análisis que avale la materia prima y el análisis científico que sustente su seguridad de uso, considerando procesos de fabricación o cualquier otro proceso que pueda alterar la biocompatibilidad del material. 50 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Copia de las publicaciones en revistas indexadas de los estudios clínicos concluyentes y con la suficiente casuística, realizados para demostrar la seguridad y eficacia del dispositivo a registrar. 2.13 Estudios clínicos (según aplique). APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Evaluación clínica que contenga el análisis exhaustivo de los datos clínicos disponibles, relativos a la finalidad de uso del dispositivo a registrar, incluyendo datos sobre su desempeño y seguridad. Deberá incluir datos específicos sobre el dispositivo a registrar, así como cualquier dato relacionado con los dispositivos que el fabricante considere comparables con el suyo. Ambos dispositivos deben tener la misma finalidad de uso y deberán ser comparados con respecto a sus características técnicas y biológicas. La evaluación deberá ser realizada por profesionales expertos en el área. Para dispositivos nuevos que no cuenten con estudios clínicos en otros países, presentar el reporte y conclusiones de estudios clínicos realizados en México, con previa autorización del protocolo de investigación, los cuales deberán ser concluyentes y con la suficiente casuística, anexar copia de las publicaciones correspondientes. Traducción completa del reporte en caso de presentarse en idioma diferente al español y/o inglés. En caso de las publicaciones, deberán presentar 3 como mínimo, siempre que sea concluyente y con la muestra poblacional suficiente para demostrar la seguridad y eficacia. Sí no hay publicaciones específicas para el dispositivo a registrar, se puede considerar una evaluación realizada por profesionales expertos en el área, mediante la cual sustenten la similitud o equivalencia con productos que ya demostraron su seguridad y eficacia, debiendo tratase de los mismos materiales, tecnología e indicación de uso del dispositivo médico a registrar. Adicional presentar las publicaciones más representativas de los Dispositivos Médicos con los que es comparable (mínimo una referencia). 51 52 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.1. Formato de solicitud 1.2. Acreditación de representante legal 1.3. Pago de derechos 1.4. Avisos de funcionamiento y de Responsable Sanitario FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 153 RIS Art. 14 LFPA Acuerdo de trámites y servicios. ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Original de solicitud Formato publicado por Cofepris para llevar a cabo la solicitud de trámites relacionados con el registro del producto. Formato vigente de autorizaciones, certificados y visitas. Debidamente llenado, los rubros que no apliquen a su solicitud deben estar cancelados con una línea paralela, firmada por el representante legal o responsable sanitario. Art. 15 LFPA Original u original de copia certificada del poder notarial o en su caso RUPA copia simple y vigente. Presentar original u original de copia certificada del Poder Notarial a favor del Representante Legal establecido en México mediante el que la empresa le confiere poderes amplios para realizar trámites de registros de los dispositivos médicos (artículo 262 LGS). Se podrá aceptar copia simple, debiendo hacer referencia al número de trámite en el cual ingreso el documento. El Representante Legal deberá ser el mismo que firme el formato de Solicitudes, o en su caso deberá ser firmada por el responsable sanitario. Art. 195-A LFD Acuerdo de trámites y servicios. Comprobante pago a. Cuota establecida por la LFD b. Para el caso de pago en banco: Sello bancario y fecha c. Impresión del pago electrónico Copia simple del aviso de funcionamiento Se presente en copia simple con los datos: a) Responsable Sanitario: b) Cédula Profesional: Formatos presentados a la Autoridad para notificar los datos del establecimiento donde físicamente se lleva a cabo una actividad relacionada con el proceso de dispositivos médicos adicional al domicilio fiscal del propio establecimiento. Copia o número de aviso de funcionamiento con los siguientes elementos o características: -Razón social de la Empresa: -Domicilio: -Clasificación Autorizada: -Líneas de Fabricación: debe incluir la del producto solicitado -Fecha de Expedición -Firmado -Número de entrada que acredita su recepción en la COFEPRIS a. El aviso debe corresponder con los datos del establecimiento asentados en el formato. b. Incluir o anexar copia simple del aviso (s) c. Los datos expresados en la clave SCIAN deben corresponder al giro del establecimiento conforme a la actividad realizada. Art. 181 RIS Art. 259 LGS NOM-241-SSA1vigente En el caso de solicitar incluir más de un distribuidor se deberán presentar los avisos de funcionamiento y de Responsable correspondientes. Con nombre y firma del propietario o representante legal. El responsable sanitario debe coincidir con la persona que avala la información técnica. El responsable sanitario deberá de cumplir con lo indicado en la norma oficial mexicana aplicable. 53 INFORMACIÓN ADMINISTRATIVA Y LEGAL FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Información completa o complementaria (contra etiqueta) en idioma español en los términos de la NOM 137 SSA1- vigente. Se pueden utilizar de manera opcional los símbolos incluidos en los apéndices normativos A e informativo B de dicha norma. 1.5. Proyectos de etiqueta o contra etiqueta Art. 179 fracc. II RIS NOM 137-SSA1vigente Proyecto de etiqueta o contraetiqueta en formato físico (copia impresa legible). Certificado de Libre Venta o equivalente expedido por la autoridad sanitaria del país de origen. Documentos equivalentes: 1.6. Certificado de Libre Venta (CLV) Art 180 fracc. I RIS Art. 153 RIS APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. 1. Carta expedida por la autoridad sanitaria del país de origen donde se indique que dichos productos no están sujetos a control sanitario. 2. Documento emitido por el Ministerio correspondiente que regule los dispositivos médicos en el país de origen. 3. Reportes de los estudios clínicos concluyentes llevados a cabo en territorio nacional de acuerdo a lo señalado en el Reglamento de la Ley General de Salud en Materia de Investigación, con protocolo de investigación previamente autorizado por esta comisión. 4. Para el caso de que el CLV no avale al fabricante legal o para la autorización del fabricante legal sin CLV, deberán presentar documento legal notariado de origen que acredite la relación entre los fabricantes como responsabilidad legal para asegurar el cumplimiento de requerimientos regulatorios y responsabilidad en la calidad del producto a registrar. En este caso, presentar también el CLV del país de origen del fabricante del producto. El proyecto de etiqueta se entrega en un tanto y en archivo de Word. Deberá incluir el listado de presentaciones con la descripción y número de código o catálogo de cada presentación del dispositivo médico (cuando aplique), que deben corresponder a los señalados en certificado de libre venta y documentación técnica/comercial presentada. Únicamente para fabricación extranjera: a. Documento legal autenticado. b. Original u original de copia certificada por notario en México. c. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses. d. Dicho documento deberá avalar: Producto (denominación distintiva y modelos), Fabricante Legal y/o Sitio de fabricación, lista de códigos de presentaciones. En caso de no incluir la lista de códigos de las presentaciones, deberá presentar carta aclaratoria emitida por el fabricante que avale dichos códigos. 54 INFORMACIÓN ADMINISTRATIVA Y LEGAL 1.7. Certificado de buenas prácticas de fabricación del (os) establecimiento (s) del fabricante (s) real (es) del (os) dispositivo médico o su documento equivalente. 1.8. Carta de representación (solo cuando el producto no es fabricado por la casa matriz, subsidiaria o filial del solicitante del registro en México). 1.9. Declaración de aval de responsable sanitario ante la COFEPRIS FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO ELEMENTO QUE INTEGRA EL REQUISITO Fabricación nacional: Oficio de Certificación de Buenas Prácticas de Fabricación emitido por la COFEPRIS. Fabricación extranjera: Certificado de Buenas Prácticas de Fabricación emitido por la autoridad sanitaria del país de origen o su documento equivalente. Art. 179 fracc. VI RIS Art. 180 fracc. III RIS Art. 153 RIS Equivalentes: •Certificado ISO 13485 versión vigente emitido por organismo autorizado. •Certificado de marca CE para dispositivos médicos emitido por organismo autorizado en la Unión Europea. •Declaración de cumplimiento de Buenas Prácticas de Fabricación incluida dentro del Certificado de Libre Venta emitido por la Autoridad Sanitaria o en su caso por el Ministerio correspondiente que regule el producto. CRITERIO Fabricación nacional: Certificado de buenas prácticas de fabricación emitido por la COFEPRIS: 1. Copia simple y que exprese: razón social, domicilio, fecha de emisión y vigencia. 2. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. Fabricación extranjera: Original u original de copia certificada de Certificado de buenas prácticas de fabricación o documento equivalente del sitio o sitios de fabricación. 1. Documento legal autenticado. 2. Documento vigente. En caso de no declararse la vigencia en el documento, la emisión del mismo no deberá ser mayor a 30 meses 3. Debe avalar la(s) línea(s) de fabricación del (los) producto(s) a registrar. 4. El Certificado ISO 13485 puede ser autenticado en el país del fabricante o país de organismo certificador. Documento emitido por el fabricante legal responsable en la cual otorga los derechos de comercialización y distribución de sus productos a una compañía establecida en territorio nacional. Art. 16 RIS Art. 180 fracc. II RIS Art. 161 fracc. III RIS Acuerdo de trámites y servicios. Art. 153 RIS Carta de representación original u original de copia certificada. En caso de solicitar incluir más de un distribuidor se deberá presentar la carta de representación para cada uno de ellos. O en su caso original de copia certificada de Carta de representación o aclaratoria en la cual un distribuidor tiene la facultad a su vez de nombrar a otros como sus distribuidores. a. Solo debe presentarse si el producto no es fabricado por la casa matriz o filial de la compañía, fábrica o laboratorio que solicite el registro en México. b. En original u original de copia certificada en original, en idioma español o traducido al español por perito traductor. c. Documento legal autenticado. d. Emitido por el fabricante legal en el extranjero. e. Otorgado a la compañía que solicita el registro en México y en su caso a otros distribuidores e importadores. En caso de ser filial, deberá presentar carta de filiales. Carta original de declaración de aval de responsable en donde avala la información técnica presentada debidamente firmada por el responsable sanitario. Y de las traducciones simples de la información técnica en caso de presentarse en idioma diferente al español o inglés. 55 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA 2.1. Información general. FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Art. 16 RIS ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Información general: 1. Denominación genérica. Documento emitido por el solicitante del registro 2. Denominación distintiva en México (en idioma español). 3. Declaración de definición: Indicación de uso, identificando: Enfermedad o afección. Situación o condición del cuidado de la salud (Crítica, seria, no seria) Descripción de la funcionalidad principal del Software como Dispositivo médico (ScDM): Características y/o funciones críticas que ayuden a establecer la significancia clínica de la información proporcionada por el ScDM (tratamiento, diagnóstico, dirigir el tratamiento clínico, informar sobre el manejo clínico) de acuerdo a la situación o condición del cuidado de la salud. La declaración de definición conducirá a una correcta clasificación con base al riesgo sanitario, de acuerdo a lo establecido en el apéndice X de la FEUM. - Clasificación de Riesgo del software como dispositivo médico. - Beneficios clínicos de la información de salida de acuerdo a la información inicial (datos de entrada). - Descripción del ScDM: Versión del ScDM Lenguaje de programación utilizado. Entorno en el que se pretende utilizar el ScDM (casa, Hospital, consultorio, clínicas). Plataforma en la cual va a ser instalado el software como dispositivo médico. Sistemas operativos con los cuales el ScDM es compatible. Intermediarios entre los resultados generados por el ScDM y el usuario: red interna, bluetooth, internet externo, internet interno. Dispositivos con los cuales va a interaccionar. Usabilidad del ScDM. Capacidad de Interoperabilidad. 56 INFORMACIÓN TÉCNICA Y CIENTÍFICA DE SEGURIDAD Y EFICACIA 2.2. Instructivo, si procede, para su uso, inserto o manual de operación FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO Art. 179 fracc. III RIS. APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. NOM 137 – SSA1- vigente Art. 16 RIS APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. 2.3. Evaluación Clínica ELEMENTO QUE INTEGRA EL REQUISITO CRITERIO Instructivo, inserto o manual de operación en formato físico (copia impresa legible) en idioma español. Deberá contener como mínimo la siguiente información dependiendo del tipo de producto: 1. Denominación distintiva y en su caso denominación genérica (conforme al Art. 2, fracción IV del RIS). 2. Versión del software. 3. Descripción. 4. Finalidad de uso. 5. Mantenimiento (cuando aplique) 6. Precauciones 7. Advertencias y leyendas alusivas correspondientes (cuando aplique) 8. Contraindicaciones (cuando aplique). 9. Incidentes adversos (cuando aplique). Validación de software como dispositivo médico, en la que se contemple la evidencia documental de las pruebas de validación analítica /técnica y validación clínica que demostrarán la seguridad, efectividad y desempeño del ScDM. De acuerdo a la indicación de uso establecida por el fabricante y a la declaración de definición. Reportes para dar cumplimiento de acuerdo a lo indicado puntos: 2.3.1. Validación analítica /técnica 2.3.2. Validación clínica Para dar cumplimiento con este punto deberá presentar: 2.3.1. Validación analítica /técnica 2.3.2. Validación clínica APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. ANEXO C. Procedimiento sugerido para la revisión de la literatura (informativo) del Suplemento para Dispositivos Médicos, FEUM. Evidencia documental emitida por el fabricante o desarrollador de las pruebas de verificación y validación del Software como dispositivo médico para garantizar el buen desempeño, calidad y seguridad. La validación analítica /técnica demuestra si el ScDM procesa correctamente los datos de entrada para generar resultados precisos, confiables y exactos. Evidencia documental emitida por el fabricante o desarrollador, de las pruebas de validación clínica del Software como dispositivo médico para demostrar el desempeño, efectividad y seguridad. La validación clínica demuestra si los datos de salida proporcionados por el ScDM (precisos, confiables y exactos) cumplen con la indicación de uso en la población objetivo. Reporte completo de las pruebas realizadas por el desarrollador o fabricante para validar y verificar el diseño, funcionalidad y desempeño del ScDM durante el ciclo de vida del mismo y demostrar que satisface las especificaciones establecidas ya sea conforme a la FEUM, u otras farmacopeas reconocidas o Normas Oficiales Mexicanas o pruebas desarrolladas conforme a especificaciones de organismos especializados (incluir referencia del estándar internacional correspondiente), u otra bibliografía científica reconocida internacionalmente. Para dar cumplimiento con este punto deberá presentar cualquiera de los siguientes documentos de evidencia clínica: a) Reporte completo del o los estudios clínicos con suficiente casuística para demostrar la seguridad y eficacia del software como dispositivo medico con resultados concluyentes. Así como su correspondiente publicación en alguna revista indexada o de reconocido prestigio médico (Aplica para el caso en que el desarrollador o fabricante requiera generar datos propios; o se trate de una nueva condición clínica; o una nueva población objetivo) 57 b) Reporte de revisión crítica en la literatura del ScDM a registrar, con la opción de utilizar o no un comparador (dispositivo médico equivalente o similar en diseño e indicación de uso), justificando la elección del mismo por parte del fabricante y de los profesionales expertos en el área. Este reporte debe contener la búsqueda sistemática de la evidencia clínica señalando criterios de inclusión y exclusión, principales fuentes de información consultadas, resumen de las publicaciones más representativas del ScDM de revistas indexadas de los estudios seleccionados para la evaluación, en los cuales se detallen los aspectos más relevantes en cuanto a metodología, discusiones, resultados y conclusiones, entre otros. c) Reporte de revisión crítica en la literatura que incluya: búsqueda sistemática y resumen de las principales publicaciones de revistas indexadas seleccionadas que plasmen los beneficios en salud del uso del ScDM, así como experiencias de los profesionales de la salud expertos en el área, señalando las principales fuentes de información consultadas. (Aplica para el caso en el cual la significancia de la información generada sea distinta al diagnóstico o tratamiento ejemplo: Modelo tridimensional generado a partir de imágenes de tomografía que sirve como simulador en la toma de decisiones de un profesional de la salud, banco de información que dirige o maneja un tratamiento). La elección de la evidencia clínica va a depender de la significancia de la información generada por el ScDM y de la situación o condición en salud, así como de la clasificación con base al riesgo sanitario: Clase I, Clase II y Clase III. En caso de que no se cuente con estudios clínicos en otros países presentar el reporte y conclusiones de estudios clínicos realizados en México, con previa autorización del protocolo de investigación, el cual deberá ser concluyente y con la suficiente casuística, anexando copia de la correspondiente publicación. Algunas medidas de validación clínica que podrán reportarse son: sensibilidad, especificidad, valores predictivos positivos, valores predictivos negativos, efectividad, ods ratio, entre otras. 58 INFORMACIÓN ADMINISTRATIVA Y LEGAL 2.4. Información del proceso del Ciclo de vida del Software como dispositivo médico FUNDAMENTO LEGAL QUE LO SUSTENTE COMO REQUISITO PARA REGISTRO CRITERIO Descripción breve o diagrama de flujo del Ciclo de vida del Software como dispositivo médico. Se puede presentar a través de: a. Resumen general del proceso de ciclo de vida del software como dispositivo médico que identifique cada uno de las actividades o fases realizadas durante el mismo. (Requisitos, especificaciones de diseño, desarrollo, pruebas de validación y verificación, mantenimiento, uso, y retiro (decommissioning). Emitido y avalado por el fabricante. 2.5. Certificado analítico o certificado de producto terminado. 2.6. Reporte de Ciberseguridad. 2.7. Reporte o informe de Gestión de riesgos. APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. ELEMENTO QUE INTEGRA EL REQUISITO Art. 179 RIS Art. 180 fracc. IV RIS NOM-241- SSA1- vigente APÉNDICE III. Normativo. Lineamientos para obtener el registro sanitario de un dispositivo médico del suplemento para dispositivos médicos de la FEUM. APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. APÉNDICE X. Normativo. Software como dispositivo médico del Suplemento para Dispositivos Médicos, FEUM. Documento emitido por el responsable de la calidad del producto o por el fabricante en donde se declaren los parámetros a evaluar y los resultados de los mismos por lote o número de serie, deberá estar emitido en hoja con membrete de la razón social del responsable de su emisión. Documento emitido por el fabricante o desarrollador del ScDM en donde se declaren los principales aspectos de Ciberseguridad. Documento emitido por desarrollador del ScDM. el fabricante o Declaración de Conformidad emitida por el fabricante o por el responsable de la calidad de la fabricación u organismo autorizado en donde se señale que el producto cumple con los requisitos establecidos para su comercialización y uso con base a normas internacionales de calidad. Consideraciones generales: Los certificados de análisis con firma electrónica se aceptarán siempre que la información declarada venga avalada por el Responsable Sanitario de la compañía que solicita el registro en México. Resumen en el cual se indiquen: a) Medidas de control en tema de ciberseguridad. b) Vulnerabilidades conocidas y previsibles. c) Proceso o mecanismos para la vigilancia de la información del ScDM. d) Evidencia de la verificación de la efectividad de los controles de seguridad debiendo contener descripción de los métodos de prueba, resultados y conclusiones. Resumen en el cual se especifiquen y se revisen los riesgos previsibles y los modos de falla que presente el ScDM durante su ciclo de vida: a) Identificación de los posibles peligros o riesgos. b) Evaluación de riesgos asociados al uso del ScDM. c) Implementación de controles para reducir los riesgos que hasta cierto nivel son aceptables. d) Evaluación de la efectividad de las medidas implementadas para la mitigación de los riesgos. e) Aspectos de seguridad (“safety” y “security”). 59 Consideraciones generales: 1. El presente documento es una guía para el armado del expediente para la obtención del registro sanitario de los insumos a que se refiere el capítulo IX, del Título Segundo del Reglamento de Insumos para la Salud, no obstante esta guía no exime del cumplimiento de requisitos técnicos y legales conforme a la legislación vigente aplicable. 2. Deberán presentar carta(s) aclaratorias emitidas por el fabricante y avaladas por el responsable sanitario, responsable legal, o solicitante del registro, cuando se requiera explicar algún punto del expediente, para subsanar discrepancias entre otras por ejemplo: nombre comercial vs nombre de diseño, cesión de derechos entre fabricante original y el fabricante actual, diferencias de razón social entre fabricación, o en su caso, cualquier diferencia en la información técnica. 3. Todos los documentos que acompañen a las solicitudes deberán presentarse en idioma español o inglés, en caso de que se presenten en otro idioma deberá adjuntarse a los mismos su respectiva traducción al español por perito traductor, avalada con la firma del responsable sanitario. (Art. 153 del Reglamento de Insumos para la Salud). 4. Se sugiere presentar la información técnica con su traducción al español, ello con la finalidad de evitar posibles correcciones internas relacionadas con la traducción de la información que se plasma en el registro sanitario otorgado. 5. En el caso de la documentación legal, se puede presentar copia simple de la copia certificada referenciando el trámite en que fue presentado el documento original u original de copia certificada, siempre y cuando el trámite al que haga referencia no tenga una antigüedad mayor a 12 meses de haberse ingresado. 6. Se sugiere que los datos que contiene el proyecto del Oficio de Registro Sanitario se presenten en formato Word editable (lista de presentaciones, formula, etc.). 7. Documento legal autenticado: a) Apostilla: Procedimiento legal que se aplica para certificar la personalidad jurídica de los firmantes garantizando la autenticidad de un documento, al cual están sujetos los países que forman parte del Acuerdo de la Convención de la Haya. b) Consularizado: Procedimiento legal que se aplica para certificar la personalidad jurídica de los firmantes garantizando la autenticidad de un documento ante el Consulado o Embajada mexicana en el país emisor del documento; se utiliza cuando el país no forma parte del Acuerdo de la Convención de la Haya o no reconoce la Apostilla como procedimiento. c) Legalizado. En el caso de países que no son miembros de la Haya y que no cuenten con Embajada o Consulado mexicano, se podrá aceptar el documento legalizado por la Autoridad Sanitaria de ese país, e incluyendo una carta aclaratoria también legalizada por la misma autoridad. Nota: Casos en los que los documentos legales pueden presentarse sin apostilla o autenticación por cónsul: 60 Si el trámite es digital: A) En caso de que el documento legal contenga una firma electrónica debe entregarse impreso o digitalizado, y éste será considerado cuando su validez y autenticidad puedan ser comprobados a través de los sitios oficiales de las Autoridades extranjeras que los emitan. El promovente debe remitir, según aplique, la ruta de acceso completa, el usuario y contraseña. B) En caso de que el documento legal sea expedido electrónicamente debe entregarse impreso o digitalizado y éste será considerado cuando su validez y autenticidad puedan ser comprobados a través de los sitios oficiales de las Autoridades extranjeras. El promovente debe remitir, según aplique, la ruta de acceso completa, el usuario y contraseña adjuntando esta información en su solicitud. Notas ScDM: 1. Para el caso en el cual el Software como dispositivo médico (ScDM) requiera del uso de un sensor o transductor necesario para medir parámetros fisiológicos (datos de entrada del software) y considerando que el fabricante es el mismo que el desarrollador del Software como dispositivo médico y que es para el uso exclusivo del software, deberá cumplir además, con los requisitos señalados en los Requisitos para el trámite de Registro Sanitario para Clase I, Clase II o Clase III, específicamente con: Descripción o diagrama de las partes funcionales del producto, materias primas, pruebas de laboratorio (considerando las pruebas de desempeño que garanticen la compatibilidad del ScDM con el sensor), información del proceso de fabricación, certificado analítico o certificado de producto terminado, esterilización para aquellos productos que ostenten la leyenda de estéril, Información de envase, estudios de estabilidad para aquellos dispositivos médicos que por sus características y finalidad de uso requieran de ostentar una fecha de caducidad y pruebas de biocompatibilidad. 2. En caso de que el Software como dispositivo médico corresponda a una aplicación móvil (App) la cual se encuentre instalada en un reloj inteligente (smartwatch) además de los lineamientos antes descritos deberá presentar reporte de pruebas de seguridad eléctrica dando cumplimiento con la norma IEC 62368-1:2020 Seguridad de los equipos electrónicos de audio/vídeo, tecnología de la información y tecnología de la comunicación y demás estándares o guías internacionales aplicables, debidamente firmado por el responsable de la emisión. 61
 0
0
Anuncio
Documentos relacionados


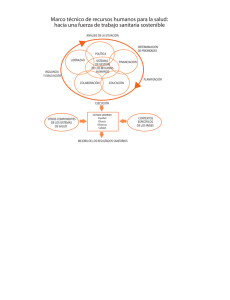
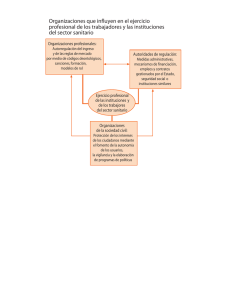
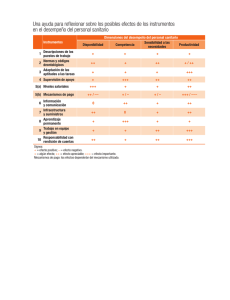



Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados